105-Bioconductor没想象的那么简单(part6)
刘小泽写于19.4.17 part1:https://www.jianshu.com/p/b030d05a80a4 part2:https://www.jianshu.com/p/c4fdab9929db part3:https://www.jianshu.com/p/f43d9d07a4af part4:https://www.jianshu.com/p/4560a29d8da2 part5:https://www.jianshu.com/p/e695b4a1dcf7 part6:https://www.jianshu.com/p/79ad7355fb8c
什么是rtracklayer?
它就是读取多种序列注释信息文件的工具,包括BED、bigwig、GTF等
比如可以在GENCODE网站下载hg38的基因组注释文件:ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_30/gencode.v30.annotation.gtf.gz
library(rtracklayer)
hg38.gencode.gtf <- import.gff(con = 'gencode.v30.annotation.gtf')
length(hg38.gencode.gtf) #2774299行数据
head(hg38.gencode.gtf) #打印前几行,还是GRange格式,注意其中gene id、transcripts id的.后内容是版本号
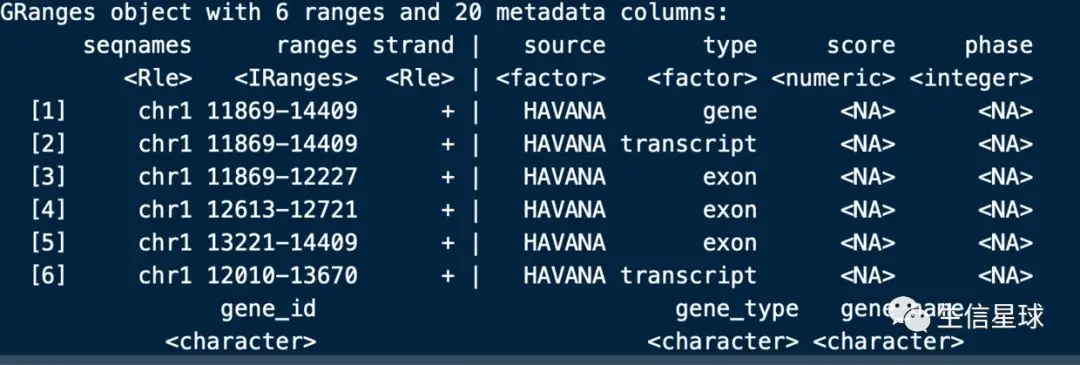
还是原来的gtf文件,和linux中不同的是,它的前三列是标准GRanges格式,后面是附加的metadata
如何进行坐标转换?
有时自己分析序列差异可能是根据hg38基因组得到的,但是同时又分析了公共数据,它是基于hg19,因此为了二者可以进行比较,需要将hg38的坐标映射到hg19上。在R中需要用到chainfile文件和liftOver函数来解决
还是先加载AnnotationHub,看看其中的数据格式
library(AnnotationHub)
ah <- AnnotationHub()
table(ah$rdataclass) #其中ChainFile是需要用到的
比如想看hg38和hg19坐标转换的文件
chain <- query(ah,c("hg19","hg38","chainfile")) # 查看相关坐标文件
chain
AnnotationHub with 2 records
# snapshotDate(): 2018-10-24
# $dataprovider: UCSC
# $species: Homo sapiens
# $rdataclass: ChainFile
# additional mcols(): taxonomyid, genome, description,
# coordinate_1_based, maintainer, rdatadateadded, preparerclass,
# tags, rdatapath, sourceurl, sourcetype
# retrieve records with, e.g., 'object[["AH14108"]]'
title
AH14108 | hg38ToHg19.over.chain.gz
AH14150 | hg19ToHg38.over.chain.gz
可以看到hg38和hg19是可以相互转换的,只需要下载需要的文件,这里我们要将hg38转为hg19
hg38.to.hg19 <- chain[["AH14108"]] # 下载hg38转hg19坐标数据
然后开始进行坐标转换
test.hg38 <- head(hg38.gencode.gtf)
test.hg38
test.hg19 <- liftOver(test.hg38,hg38.to.hg19) # 转换过程,先放原始数据,再放转换坐标文件
test.hg19 #得到的是一个GRangesList
new.test.hg19 <- unlist(test.hg19)
如何从GEO中下载大量的原始数据或者表达量数据?
一般一个实验会涉及十几或者几十样本,更夸张的,像单细胞数据,成白上千个单个细胞样本数据存在一个GEO ID中,虽然每个细胞的数据量不大,但是这么多如何才能下载?
以18年12月的利用smart-seq2进行的成纤维单细胞CAFs分析文章为例: Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing
它的数据存在了 GSE111229,其中有768个sample
library(GEOquery)
test.geo <- getGEO("GSE111229")
下载的这个是一个列表,用length(test.geo) 看一下就知道,结果是1,现在取出真正的数据,也就是一个ExpressionSet
> test.data <- test.geo[[1]]
ExpressionSet (storageMode: lockedEnvironment)
assayData: 0 features, 768 samples
element names: exprs
protocolData: none
phenoData
sampleNames: GSM3025845 GSM3025846 ... GSM3026612 (768 total)
varLabels: title geo_accession ... tissue:ch1 (47 total)
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
pubMedIds: 30514914
Annotation: GPL13112
然后expressionSet有很多参数,像上面的phenoData就可以用pData()来获取,可以用sampleNames获得样本名
pd <- pData(test.data)
sampleNames(test.data)
pd是一个数据框,其中有一列taxid是物种分类号:10090,表示物种是小鼠;另外还有data_processing的信息,包括比对、使用RPKM定量等;再往后,relation.1中记录了每个样本的SRA位置,可以下载;有的GSE数据还给出了每个样本的表达量数据(一般在supplement data中),那么就可以$直接提取那一列,as.data.frame()保存为数据框,write.csv()输出为文本文件,最后用循环下载wget -i data.txt