144-重温那些好用的R小知识
刘小泽写于19.10.30 经常使用R,势必会遇到一些好用的小函数,帮忙处理数据,这里就做一下整理
数据读取与基本操作
首先需要用到一个数据,来自:https://raw.githubusercontent.com/Bioconductor/BiocWorkshops/master/100_Morgan_RBiocForAll/ALL-phenoData.csv
数据长这样
一共4列:样本(一个特有的ID号)、性别、年龄、每个病人的细胞特征(例如BCR/ABL表示存在BCR/ABL易位、NEG表示没有明显的细胞特征)

读入数据
先找到这个文件,它会弹出一个对话框,然后找到文件后,这个fname就保存了文件的路径
fname <- file.choose()
fname
检查数据是否存在
file.exists(fname)
然后读取并查看数据
pdata <- read.csv(fname)
> head(pdata)
Sample sex age mol.biol
1 01005 M 53 BCR/ABL
2 01010 M 19 NEG
3 03002 F 52 BCR/ABL
4 04006 M 38 ALL1/AF4
5 04007 M 57 NEG
6 04008 M 17 NEG
> dim(pdata)
[1] 128 4
> summary(pdata)
Sample sex age mol.biol
01003 : 1 F :42 Min. : 5.00 ALL1/AF4:10
01005 : 1 M :83 1st Qu.:19.00 BCR/ABL :37
01007 : 1 NA's: 3 Median :29.00 E2A/PBX1: 5
01010 : 1 Mean :32.37 NEG :74
02020 : 1 3rd Qu.:45.50 NUP-98 : 1
03002 : 1 Max. :58.00 p15/p16 : 1
(Other):122 NA's :5
> class(pdata)
[1] "data.frame"
可以在读取同时指定每一列类型
# 之前读入的数据第一列ID,它的数据类型默认是因子型,但是我们只想要字符
> class(pdata$Sample)
[1] "factor"
# 因此可以在读入数据的时候,自己修改
pdata2 <- read.csv(fname,
colClasses = c('character','factor','integer','factor'))
> class(pdata2$Sample)
[1] "character"
取子集
可以根据名称、位置、逻辑判断三种方法
# 名称
pdata[1:5, c("sex", "mol.biol")]
# 位置
pdata[1:5, c(2, 3)]
# 逻辑
subset(pdata, sex == "F" & age > 50)
bcrabl <- subset(pdata, mol.biol %in% c("BCR/ABL", "NEG"))
查看并修改因子型
有时得到的数据会存在多个因子,但有的因子是空的,我们可以把这些空的因子去掉
> table(bcrabl$mol.biol)
ALL1/AF4 BCR/ABL E2A/PBX1 NEG NUP-98 p15/p16
0 37 0 74 0 0
# 现在就可以只保留有数据的因子
factor(bcrabl$mol.biol)
bcrabl$mol.biol <- factor(bcrabl$mol.biol)
> table(bcrabl$mol.biol)
BCR/ABL NEG
37 74
使用公式探索数据
公式符号一般是:lhs ~ rhs,左手边(left hand side, lhs)是因变量(也就是Y轴);右手边是自变量
例如探索不同细胞类型的病人年龄差异,那么自变量就是不同的细胞类型病人(因子型向量),因变量就是年龄(数值型向量)
boxplot(age ~ mol.biol, bcrabl)
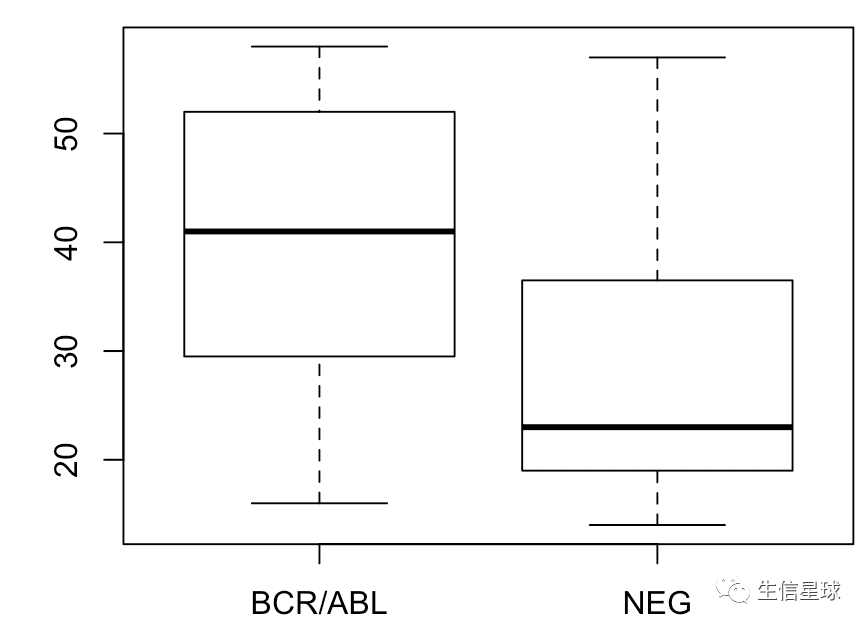
图中可以看到,BRC/ABL这一组的年龄要比NEG组的高,那么再验证一下:
# 看到差异是非常显著的
> t.test(age ~ mol.biol, bcrabl)
Welch Two Sample t-test
data: age by mol.biol
t = 4.8172, df = 68.529, p-value = 8.401e-06
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
7.13507 17.22408
sample estimates:
mean in group BCR/ABL mean in group NEG
40.25000 28.07042
不过,箱线图展示的是:最小值、第一四分位数、中位数、第三四分位数与最大值,并没有平均数,我们其实可以自己加上去统计量
正好前几天看到Y叔的推送,学习了一下
library(ggplot2)
d <- data.frame(age=na.omit(bcrabl)$age, type=na.omit(bcrabl)$mol.biol)
> head(d)
age type
1 53 BCR/ABL
2 19 NEG
3 52 BCR/ABL
4 57 NEG
5 17 NEG
6 18 NEG
# 散点图
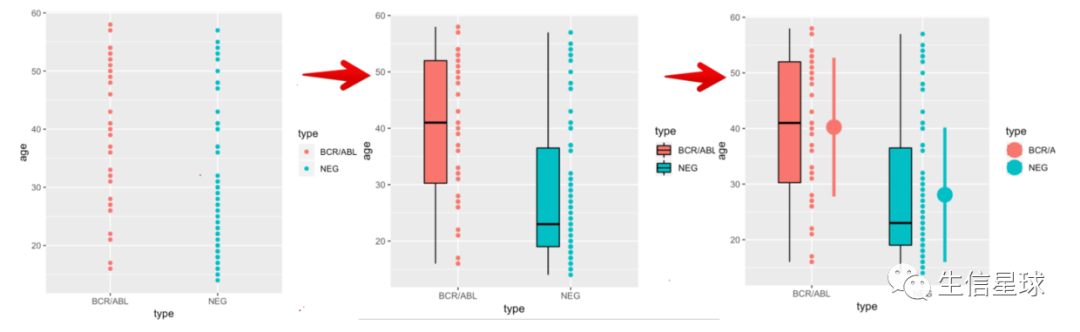
ggplot(d,aes(type,age,colour=type)) + geom_point()
# 箱线图(只有四分位数)
ggplot(d,aes(type,age,colour=type)) + geom_point() +
geom_boxplot(aes(fill=type),colour='black',width=.2,
position = position_nudge(x=-.2))
# 再添加其他统计值(如平均数和标准差)
# 附:这个符号%>% 可以用快捷键 shift+cmd+M
library(tidyverse)
ggplot(d,aes(type,age,colour=type)) + geom_point() +
geom_boxplot(aes(fill=type),colour='black',width=.2,
position = position_nudge(x=-.2))+
geom_pointrange(data=function(d){
group_by(d,type) %>%
summarize(mean=mean(age),sd=sd(age))
}, aes(y=mean, ymin=mean-sd,ymax=mean+sd),
position=position_nudge(x=.2),size=1.5)
Bioconductor数据结构相关
Bioconductor目前存在接近3000个R包,访问:https://bioconductor.org/packages/release/BiocViews.html#___Software
主要有这么四类:
- Software:分析相关
- Annotation:注释相关,例如基因名转换、富集分析数据库、各种基因组坐标对应关系
- Experiment:高质量的实验数据,例如转录组学习常用的
airway包、TCGA数据curatedTCGAData - Workflow:流程学习
利用正则表达式匹配R包
> BiocManager::available("TxDb.Hsapiens")
[1] "TxDb.Hsapiens.BioMart.igis"
[2] "TxDb.Hsapiens.UCSC.hg18.knownGene"
[3] "TxDb.Hsapiens.UCSC.hg19.knownGene"
[4] "TxDb.Hsapiens.UCSC.hg19.lincRNAsTranscripts"
[5] "TxDb.Hsapiens.UCSC.hg38.knownGene"
查看帮助文档
browseVignettes("simpleSingleCell")
三个重要的R包~ 第一个:rtracklayer
提供了import()函数,能够读入多种格式(BED, GTF, VCF, FASTA)等。下面将使用数据:https://raw.githubusercontent.com/Bioconductor/BiocWorkshops/master/100_Morgan_RBiocForAll/CpGislands.Hsapiens.hg38.UCSC.bed
suppressMessages(library("rtracklayer"))
fname <- file.choose()
fname
file.exists(fname)
发现下载的文件是.bed.txt结尾,而import()函数只识别.bed。因此需要重命名,去掉后缀.txt
ori_name <- str_split(fname,'\\/',simplify = T)[10]
aft_name <- gsub('.txt','',ori_name)
file.rename(ori_name,aft_name)
读入后是GRanges对象
cpg <- import(aft_name)
> head(cpg)
GRanges object with 6 ranges and 1 metadata column:
seqnames ranges strand | name
<Rle> <IRanges> <Rle> | <character>
[1] chr1 155188537-155192004 * | CpG:_361
[2] chr1 2226774-2229734 * | CpG:_366
[3] chr1 36306230-36307408 * | CpG:_110
[4] chr1 47708823-47710847 * | CpG:_164
[5] chr1 53737730-53739637 * | CpG:_221
[6] chr1 144179072-144179313 * | CpG:_20
-------
seqinfo: 254 sequences from an unspecified genome; no seqlengths
标准BED文件是0-based,半开半闭区间,比如[0,10) 就是 0..9 十个数,而import()函数自动将BED的类型转成Bioconductor统一类型:1-based、全闭区间,也就是将[0,10)变成了[1,10],还是原来的十个数值
三个重要的R包~ 第二个:GenomicRanges
对基因组坐标区域操作,如exons, genes, ChIP peaks, called variants
只对标准的染色体1-22+X+Y进行处理
cpg <- keepStandardChromosomes(cpg, pruning.mode = "coarse")
> head( start(cpg) )
[1] 155188537 2226774 36306230 47708823 53737730 144179072
> head( cpg$name )
[1] "CpG:_361" "CpG:_366" "CpG:_110" "CpG:_164" "CpG:_221" "CpG:_20"
取子集
subset(cpg, seqnames %in% c("chr1", "chr2"))
取两个GRanges对象的交集
这里还需要用到一个R包:TxDb开头的注释包,其中包含外显子、基因、转录本的坐标
library("TxDb.Hsapiens.UCSC.hg38.knownGene")
tx <- transcripts(TxDb.Hsapiens.UCSC.hg38.knownGene)
# 取外显子坐标:exons(TxDb.Hsapiens.UCSC.hg38.knownGene)
# 取基因坐标:genes(TxDb.Hsapiens.UCSC.hg38.knownGene)
tx
# 同样也是保留标准染色体信息
tx <- keepStandardChromosomes(tx, pruning.mode="coarse")
tx
# 统计每个转录本上有多少CpG位点
olaps <- countOverlaps(tx, cpg)
length(olaps)
table(olaps)
# 添加结果到GRanges
tx$cpgOverlaps <- countOverlaps(tx, cpg)
tx
# 根据这个结果取子集
subset(tx, cpgOverlaps > 10)
三个重要的R包~ 第三个:SummarizedExperiment
SummarizedExperiment 它既是一个包,也是一个对象。
这个对象有行和列两个维度
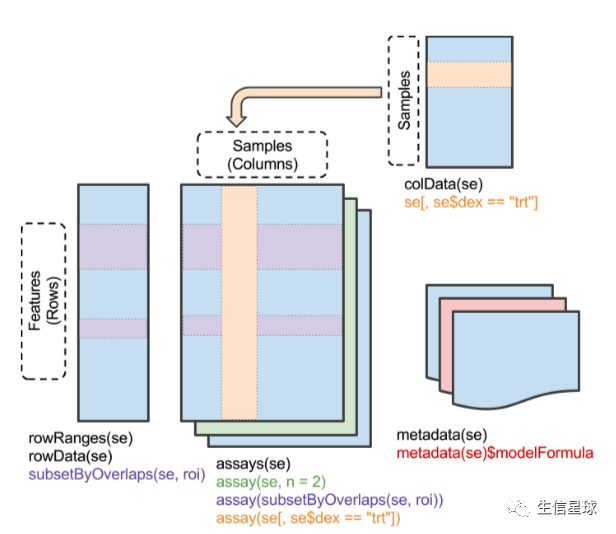
需要矩阵而不是数据框(下面只是演示方法):
library("SummarizedExperiment")
counts <- as.matrix(counts)
se <- SummarizedExperiment(assay = counts, colData = colData)
# 取子集
subset(se, , dex == "trt")
# 新增colData数据
se$lib.size <- colSums(assay(se))
colData(se)
DESeq2就是利用的SummarizedExperiment 中的count data信息;另外需要一个实验设计矩阵,例如将colData中的cell作为协变量,dex药物处理作为主要影响因子
library("DESeq2")
dds <- DESeqDataSet(se, design = ~ cell + dex)
# 如果直接根据矩阵构建对象
# dds <- DESeqDataSetFromMatrix(
# countData = countData,
# colData = colData,
# design = ~ condition)
dds <- DESeq(dds)
res <- results(dds)
resOrdered <- res[order(res$padj),]
Bioconductor注释信息相关
都有哪些注释?
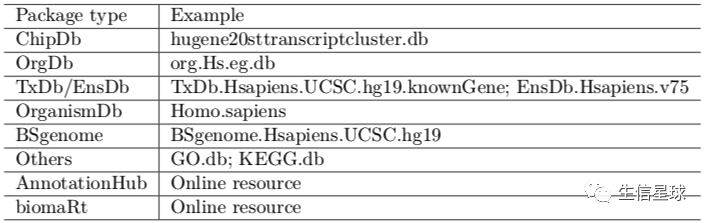
如何操作各种注释包?
例1:芯片ID转基因Symbol
固定的select(annopkg, keys, columns, keytype)
library(hugene20sttranscriptcluster.db)
set.seed(12345)
ids <- featureNames(eset)[sample(1:25000, 5)]
ids
#> [1] "16908472" "16962185" "16920686" "16965513" "16819952"
select(hugene20sttranscriptcluster.db, ids, "SYMBOL")
#> 'select()' returned 1:1 mapping between keys and columns
#> PROBEID SYMBOL
#> 1 16908472 LINC01494
#> 2 16962185 ALG3
#> 3 16920686 <NA>
#> 4 16965513 <NA>
#> 5 16819952 CBFB
最后的这个keytype不指定的话,就是默认的central ID。例如芯片数据的keytype就是探针ID;物种注释(如org.Hs.eg.db)的keytype就是Entrez ID。一个数据库那么如何获得keytype信息呢?
keytypes(hugene20sttranscriptcluster.db)
# 或columns(hugene20sttranscriptcluster.db)
[1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT"
[5] "ENSEMBLTRANS" "ENTREZID" "ENZYME" "EVIDENCE"
[9] "EVIDENCEALL" "GENENAME" "GO" "GOALL"
[13] "IPI" "MAP" "OMIM" "ONTOLOGY"
[17] "ONTOLOGYALL" "PATH" "PFAM" "PMID"
[21] "PROBEID" "PROSITE" "REFSEQ" "SYMBOL"
[25] "UCSCKG" "UNIGENE" "UNIPROT"
除了上面的select,还有一个类似的mapIds:
ids <- c('16737401','16657436' ,'16678303')
mapIds(hugene20sttranscriptcluster.db, ids, "SYMBOL", "PROBEID", multiVals = "list")
mapIds(hugene20sttranscriptcluster.db, ids, "SYMBOL", "PROBEID", multiVals = "CharacterList")
# 去掉没有匹配上的
mapIds(hugene20sttranscriptcluster.db, ids, "SYMBOL", "PROBEID", multiVals = "filter")
# 将没有匹配上的标记为NA
mapIds(hugene20sttranscriptcluster.db, ids, "SYMBOL", "PROBEID", multiVals = "asNA")
例2:转录本注释包
一般的写法是:TxDb.Species.Source.Build.Table,例如:TxDb.Hsapiens.UCSC.hg19.knownGene
select(TxDb.Hsapiens.UCSC.hg19.knownGene, c("1","10"), c("TXNAME","TXCHROM","TXSTART","TXEND"), "GENEID")
例3:位置注释
利用GRanges或者GRangesList
gns <- genes(TxDb.Hsapiens.UCSC.hg19.knownGene)
# 把每个基因的所有转录本全部提取出来,每个基因是列表中的元素,然后每个基因下面可能又包含多个转录本
txs <- transcriptsBy(TxDb.Hsapiens.UCSC.hg19.knownGene)