017-一个神奇的小软件bioawk
刘小泽写于18.7.29
写在前面:之前介绍过文本处理的三剑客,grep、sed、awk,也曾说过,学了awk,让一切字符秒变尴尬“awk word”。其中的awk可以说是独当一面了,他自己本身就是一门编程语言
bioawk是什么
先说一个生信牛人——李恒,废话不多说,一张图便知。李恒的文章随随便便就是几千的引用。最高的两篇引用上万次的,分别是BWA和SAMtools对应的文章。曾发过的nature,他是一作,另一个是Sanger实验室的合作伙伴,只有俩人,就是这么任性!他还经常为生信界的旗舰刊物Bioinformatics投稿,好像是为了**“拯救”** 这个旗舰级却只有7分的期刊。至于他的爱好嘛~就是让别人都知道他的名字

曾经在github上有讨论awk的使用,做生信的人都反映awk是很好,但用起来总有那么一点不顺手,毕竟不是为生信而生嘛!
李恒看到了,拿来awk的源代码修修补补,最终bioawk诞生
bioawk能干啥
fasta/fastq文件快速获得其中的gene id、gc含量等信息-》它一行命令搞定
在通用文件格式中,设置新的变量代表某一列或某一行 例如,在读取sam文件中,结果会有一列CIGAR,就像这里的
30S71M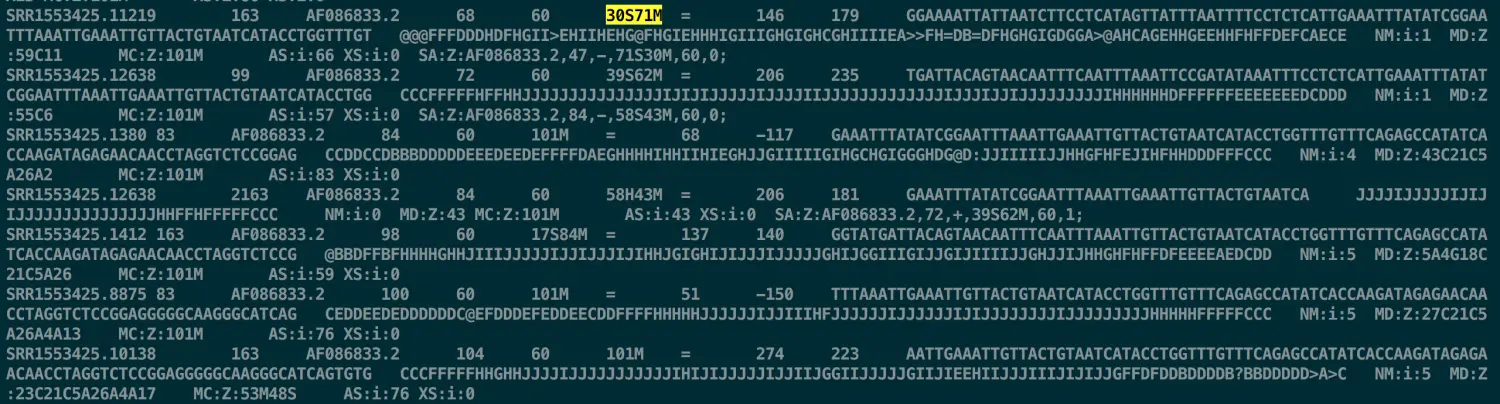
记录了位点的详细信息,比如M代表比对上(match),I代表插入(Insertion),D代表缺失(Deletion)等;如果用到这些信息,想把这一列提取出来的话,用传统的办法就是记住CIGAR在第六列,然后提取,但是这个文件有12列,不会时刻记得那么清楚。 于是,bioawk中有个
$cigar变量,直接提取出来你想要的增加和生信相关的一些函数,例如:
revcomp直接获取反向互补序列
bioawk怎么用
安装
推荐使用conda安装。一行命令就搞定,但一定一定要注意,conda的使用要用-p指定安装目录,使conda安装和自己手动安装的文件保持分离,这样防止conda随意改变你已经配置好的其他变量。
如果还没有conda,可以参考我之前介绍过的conda的使用
安装命令: connda install -p ~/YOUR/conda path/ bioawk -y
参数设置
-c指定文件格式:常用的有bed、sam、vcf、gff、fastax,甚至文件的注释行header也可以【指定header后,空格和特殊字符将会被下划线替代】

这是bioawk针对不同文件内置的变量,和源文件格式对应
【小提示:如果日后忘了文件的具体格式,可以通过bioawk -c help查看】
-t 将分隔符设为tab
-H 输出文件中依然保留注释头信息(retain header in such SAM files)
快速上手
处理FASTA文件
注释行用
$name表示,序列行用$seq表示统计序列长度
bioawk -c fastx '{print $name, length($seq)}' input.fa结果得到 chrX 155270560
统计GC含量
bioawk -c fastx '{print $name, gc($seq)}' input.fa结果得到 chrX 0.384356
得到反向互补序列
bioawk -c fastx '{print ">"$name;print revcomp($seq)}' input.fa输出长度大于100bp的序列
bioawk -c fastx 'length($seq)>100 {print ">"$name;print $seq}' input.fa然后,在后面加一个
| wc -l就能统计长于100bp的序列个数啦以后再遇到像:几百上千条序列,求其中长度大于几百bp的序列个数这样的问题,心里就不会再慌慌了
在序列注释行首或行尾添加注释
首行首添加,PREFIX可以替换成其他内容
bioawk -c fastx '{print ">PREFIX"$name; $seq}' input.fa首行行尾添加,SUFFIX可以替换成其他内容
bioawk -c fastx '{print ">"$name"|SUFFIX";$seq}'input.fa有时需要将fa中的注释行和序列行分开处理
将注释行和序列行添加分隔符tab即可,这样打开就是表格形式,注释行在第一列,序列行在第二列,导出为excel都不怕
bioawk -t -c fastx 'print $name,$seq' input.fa处理fastq文件
还是使用
-c,程序会自动识别fq文件,然后再fa两个变量$name、$seq的基础上,再添加两个$qual、$comment统计reads条数
bioawk -t -c fastx 'END {print NR}' input.fastqfq转fa
bioawk -c fastx '{print ">"$name; print $seq}' input.fastq好简单的一条命令,用perl写脚本处理要好多行,下面👇是我用perl写的

得到fq平均Phred质量值
bioawk -c fastx '{print ">"name; print meanqual($qual)}' input.fastqmeanqual是求平均质量值的函数,这个都定义好了
过滤掉短于10bp的reads
bioawk -c fastx 'length($seq) > 10 {print "@"$name"\n"$seq"\n+\n"$qual}' input.fastq注意双引号引用的值是
@、\n、\n+\n,不加引号就把这些字符直接打印出来了要根据序列质量值来剪掉trim某部分序列
bioawk -c fastx 'trimq(30,0,5){print $0}' input.fastq意思是剪掉质量值低于30,碱基位置从0-5的片段
处理BED文件
求feature信息的长度
bioawk -c bed '{print $end - $start}' test.bed处理SAM文件
将未比对上的(unmapped)序列提取出来
bioawk -c sam 'and($flag,4)' input.samflag 的值为4代表未比对上
将比对上的(mapped)序列提取出来
bioawk -c sam -H '!and($flag,4)' input.sam根据sam文件创建fasta
bioawk -c sam '{ s=$seq; if(and($flag,16)){s=revcomp($seq)} print ">"$qname“\n”s' input.sam > out.fasta解释一下:sam文件中的序列有两种情况:正向序列和反向序列。如果是反向序列就需要用 revcomp函数转换,在sam文件中这个函数只会把序列反向,不会将它互补。判断是否为反向序列就看flag信息,flag=16为反向序列。我们先把序列信息赋值给s;然后再判断是否为反向序列,是的话用revcomp处理一下再赋值给s。最后print,注意大于号和\n要加双引号
处理自定义文件
抓取某些特定条件,比如这列年龄大于20
name age hobby phone harry 12 bioinfo 1234 sam 24 bioinfo 4566 bioawk -t -c header '$age > "20" {print $0}' input.txt