161-用了这么久的bowtie2,却不知道它的结果含义?
刘小泽写于2020.2.4 经常出现这么一种情况,为了完成目标,一定会先快速做一遍,然后得到最直接的结果。等后来就会发现,原来其中还有很多不知道的事情
这次就会以bowtie2的结果为例,来说说为什么会有这种感受
首先还是先回顾下bowtie2的基本知识吧
它的官网在:http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml
一句话概括这款软件:
Bowtie 2 is an ultrafast and memory-efficient tool for aligning sequencing reads to long reference sequences.
关于使用
使用它首先需要对参考基因组构建索引
【注意这里是基因组而不是参考转录组,因为它的BWA一样,是为基因组测序而生;如果是要比对参考转录组,那么就考虑hisat2吧】
关于索引有两种获得方法:
官网下载:提供了人、小鼠、大鼠的索引下载
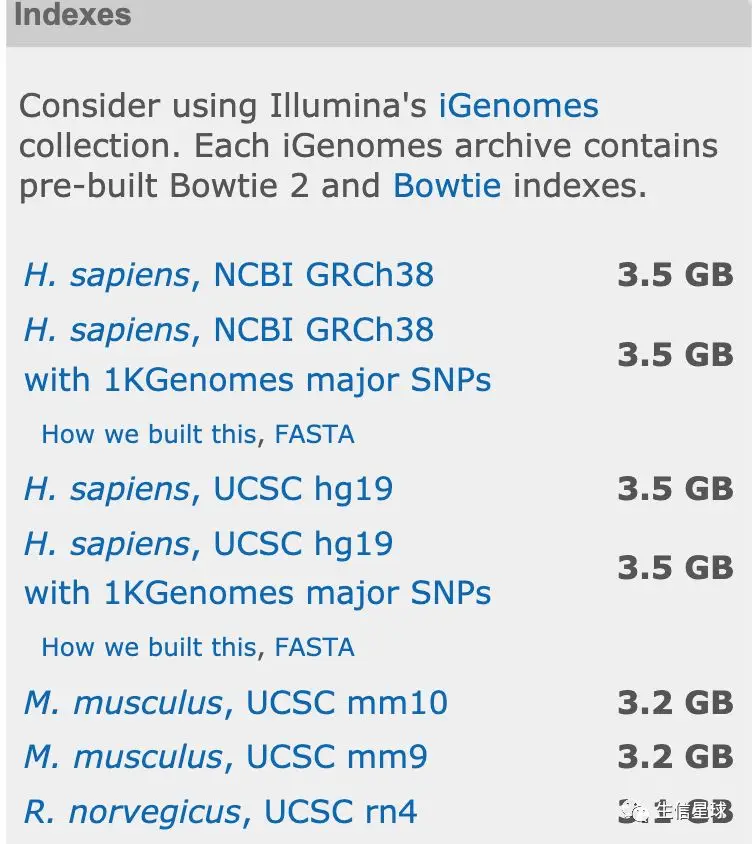
自己构建:其实自己下载好参考基因组后,自己构建也是很方便的。只需要指定三个参数:一个线程数,一个参考基因组的位置,最后就是输出的索引前缀【一个小技巧就是输出的前缀直接用物种的缩写,比如mm10、hg19、ath等】
# 举个例子 ref=/home/genome/genome.fa bowtie2-build --threads 5 $ref hg19
有了参考索引,就能进行单端或者双端的比对
可能你会想,现在都是二代PE测序了,为什么还有单端的存在呢?但如果接触了Chipseq数据,就能看到大量的单端测序
# 单端,其中 -p指定线程数
index=/home/genome/hg19
fq=/YOUR_PATH/
bowtie2 -p 5 -x $index -U $fq
# 双端
index=/home/genome/hg19
fq1=/YOUR_PATH/
fq2=/YOUR_PATH/
bowtie2 -p 5 -x $index -1 $fq1 -2 $fq2
必须注意的是,bowtie2 -x这里一定要写好路径,并且是写到前缀,于是可以看到index这个变量就指定到了hg19这里
既然是第二代,那么它的第一代有啥区别?
之前其实一直没有考虑过这个问题,因为大家都用bowtie2,而且各种文章也是用新的版本。就是“喜新厌旧”吗?其实这些在官网都有说过,只不过没有特殊情况,我们一般也不会去看,因为很多的中文教程足够使用。
第一代版本发布于2009年,当时二代测序还没有兴盛,因此它的目标是比对20-50bp的短reads。但是后来随着测序通量(每天测序得到的碱基数更多)和读长的增大(每条read能测到的长度更长),bowtie1不能满足日益发展的比对需求,才有了二代。
主要的区别有:
- 对于长度大于50bp的reads,bowtie2的速度更快,更灵敏,使用的内存也少;但如果小于50bp,依然是bowtie1更快
- bowtie1只寻找没有gap的比对,而bowtie2支持了跨gap的比对,gap的数量和长度都没有限制
- bowtie2支持局部比对(local alignment)和双端比对(end-to-end alignment),双端比对这一点和bowtie1一样
- bowtie2对read长度没有限制,而bowtie1最长1000bp
- bowtie2允许比对参考基因组的未知碱基(N),而bowtie1不可以
- 对于双端测序的reads比对,bowtie2更灵活,因此一会在结果解释中会看到,双端测序的合理比对和不合理比对
重点是结果如何解读
以往都是关注最后一行,也就是最后计算出来的比对率,看着差不多也就得了
看看样子
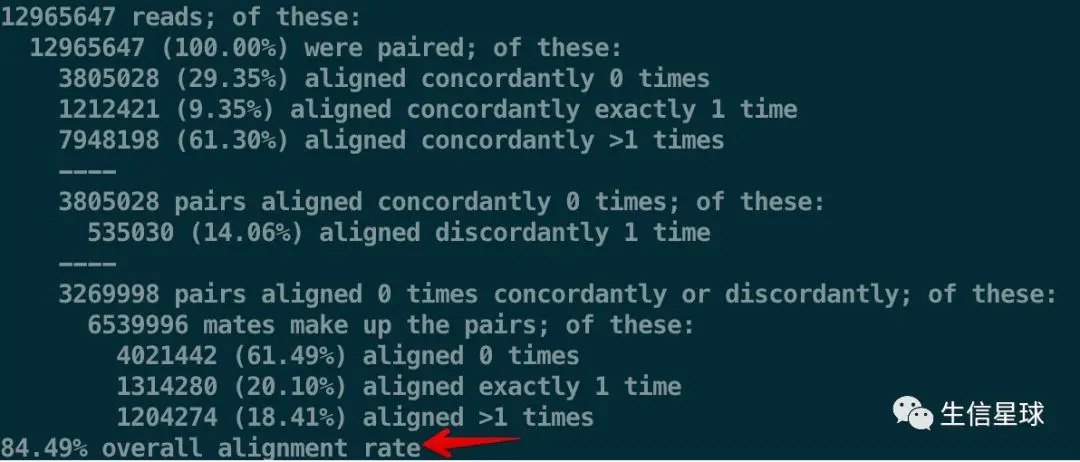
双端结果如下:
单端结果如下:

需要注意的是,这些具体的比对信息,bowtie2将他存储在了标准错误输出中,也就是说我们需要指定2>align.log这样来保存结果
逐步解读
就以上面👆双端结果为例 其中以
----为分割线,分成了三大部分
第一行:说明一共有12965647对reads进行了比对【注意这里因为是PE测序,所以是一对一对的,如果论条的话,就应该乘以2】
接着,重点关注一个名词:aligned concordantly,直译就是比对结果是不是合理。如果read1和read2都同时比对上,并且比对后的结果也符合逻辑,那么就是合理的;如果read1和read2能同时比对上,但它们各自比对的位置和本身read1、2之间的间隔差距太大,或者它们比对的方向压根就是一样的,这样就是不合理(因为本身测序得到的read1和read2应该比对到不同的链)
分割的第一部分:在共有12965647对reads中
- 3805028 (29.35%)对reads没有合理的比对
- 1212421 (9.35%)对reads合理比对了一次
- 7948198 (61.30%)对reads合理比对了多次
分割的第二部分:在没有合理比对的3805028对reads中
- 535030 (14.06%)对reads 是双端比对但不合理
因此这里要注意了:不合理比对不代表这对reads不重要,它包含了三种情况:都比对上但比对不合理(就是这里的535030)、两个read都没比对上、只有一个read比对上
分割的第三部分:在不合理比对的3805028对reads中,除去双端比对但不合理的535030对,还剩余6539996条(这里没有写3269998对,是因为这些read可能就不是配对的了),可以看图中第三部分使用的词语是mates而不是paired,mates在这里指的就是由不同read组成的群体
- 4021442 (61.49%) 条一次没比对上
- 1314280 (20.10%) 条比对上一次
- 1204274 (18.41%) 条比对上多次
最后计算:84.49% overall alignment rate
如何计算? 其实也就是统计了比对上的条数:
双端比对的结果(比对1次及多次,注意要乘以2)+ 单端比对的结果(比对1次及多次)
# 来自分割的第一部分
1212421 x 2 + 7948198 x 2
# 来自分割的第二部分
535030 x 2
# 来自分割的第三部分
1314280 + 1204274
# 最后总共比对的条数是
21909852
# 全部条数是
12965647 x 2 = 25931294
# 最后的比例就是
21909852/25931294*100%=84.49%
补充
- bowtie2默认会同时寻找
concordant和discordant的比对,除非设置--no-discordant - 和bowtie2类似,举一反三,hisat2的结果也是这么理解