168-跟着刘小泽一起回顾标准化方法
刘小泽写于2020.2.20 常见的差异分析第一步往往是去进行标准化处理,那么究竟标准化的对象是什么?怎么根据具体情况去标准化呢?这是难点所在!
首先要熟悉表达矩阵
表达矩阵就是我们上游分析根据比对后的sam/bam文件,利用软件(如转录组的featureCounts)定量的结果。它的格式是:行为基因,列为样本,中间的数值是表达量【例如下图】
标准化需要考虑的三个层面和三个因素
「三个层面」之一:组间样本间比较(两个样本两个组)
**它探讨的问题就是:**我们想看经过处理后样本的样本变化,原来我这个基因的表达量是10,那么处理后样本中这个基因表达量会下降还是上升或者不变呢?。一般我们希望这个结果有很明显的差异【也就是常说的差异分析】
「三个层面」之二:组内重复样本间比较(两个样本一个组)
**它探讨的问题就是:**单纯看看处理组的三个重复之间,某个基因有没有差别,好检测一下做的实验误差大不大。一般我们不希望这个结果很明显的差异
「三个层面」之三:组内样本内比较(一个样本一个组)
**它探讨的问题就是:**我这个组的一个重复样本中的A基因和B基因,哪个表达量更高一些?为什么对于我这个样本,B基因表达量会高于A呢?
首先要了解实验属于哪个层面,然后有的放矢,再去看应该考虑哪些情况
「三种情况」之一:测序深度
如果要比较两个样本的同一个基因表达量,那么首先要保证外部因素一致,最直接的体现就是测序深度。不同批次的测序,它的测序深度可能不同,因此可能导致下面这种情况:

【**注意:**图中所有红色和绿色的小长条就是比对到基因的测序read,我们也是按它们的数量来判断某个基因表达量的;看到有的长条之间连了一条虚线,这表示它是跨越内含子比对的测序read。我们使用的hisat2就支持这种跨内含子比对】
**图中:**貌似A样本的所有基因表达量都是B的两倍。但如果A样本本身测序量就大呢?也就是说,A和B的“家底”就不同,不能直接放一起比较,要比也要放在同一水平公正去比
「三种情况」之二:基因长度
如果要比较同一个样本中的不同基因,那么就要处理好“内部矛盾”——基因长度的问题。基因的长度是固定的,如果要比较的两个基因中,一个基因本身就很长,那么理所应当,落在上面的reads数量应该也越多。因此可能导致下面这种情况:
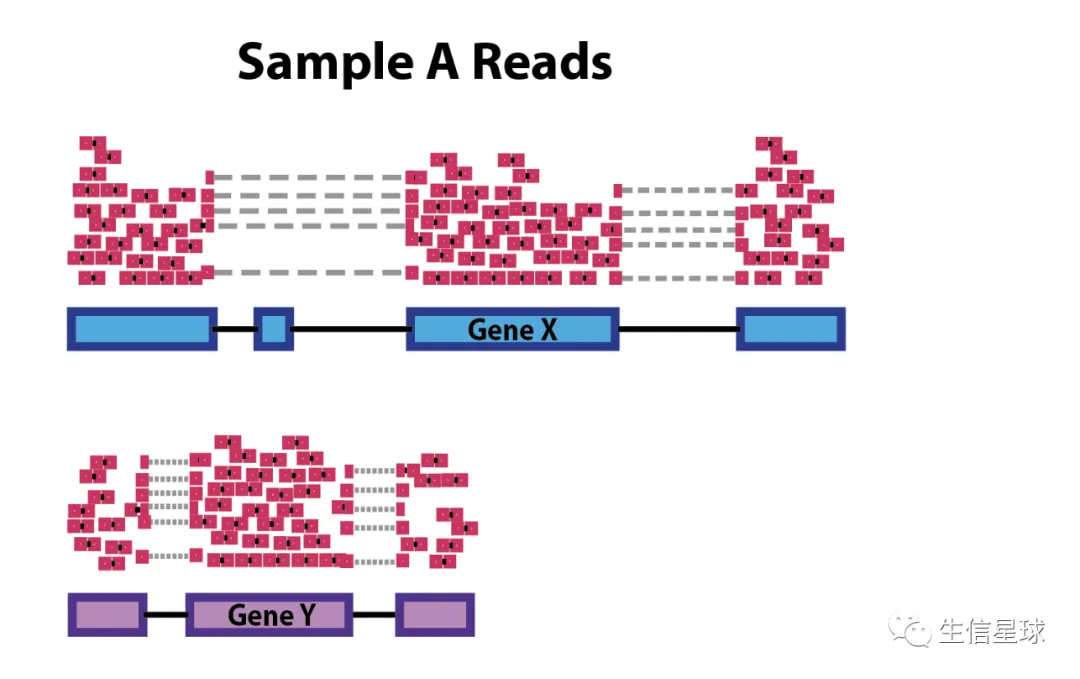
图中: 虽然都位于样本A,但基因X比Y要长,所以比对过程中也有更多的reads落在X上,这样X的获胜很有可能是靠着自己“修长”的身材,而不是靠真正的表达量实力
「三种情况」之三:RNA组成
这种情况也可以理解为:找出搅局者。依然是在组间比较,但可能某个样本中就是存在这样的**“异类”:它本身很强大,表达量超级高,拉高了整体的表达量,造成一种“虚假繁荣”**。于是当默认按照情况一:测序深度处理时,“异类”的所有小伙伴都要陪着它一起缩小,为了减小所谓的“测序深度影响”。但它的小伙伴亏啊,本来和对照样本中的基因单挑是完胜的,但这一缩减,就不行了【如下图】
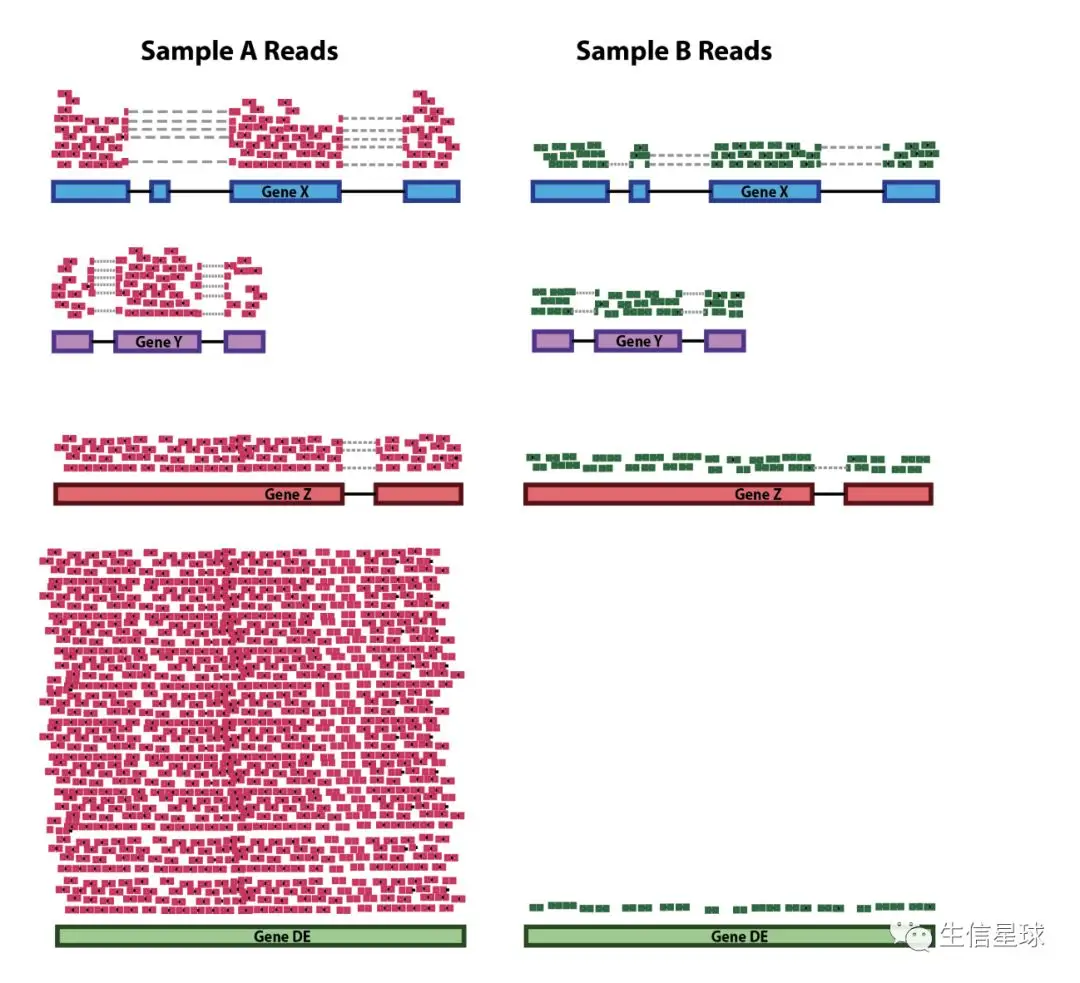
图中: 本来样本A中的所有基因都比B要高,即使为了公平起见处理一下测序深度,也很有可能是胜出或者打个平手。但A中出现了一个DE基因(可以理解成表达迥异的基因),它的到来让我们误以为样本A的测序深度很高,于是给A的所有基因都除以了一个值,保证和样本B可比。于是,样本A中除了DE以外的基因,最后都被B的对应基因打败了【A中基因含恨而亡,DE很愧疚】
但你说,这个DE基因怎么来的呢?天生的吗?
有可能是真的存在这种少数特殊基因,就在某个样本中高表达;但还有可能,是出现了样本污染混入的其他基因,它本身不属于样本A,只是过来跑跑龙套,然后带偏了整个结果
因此,在我们进行差异分析时,除了考虑测序深度,还有考虑样本内部组成
好,来看看常见的标准化方法
既然上面👆我们了解了为什么进行标准化以及标准化针对的对象,那么接下来就看看都有什么好办法来完成这个任务,以及它们适用的场景吧!
这里必须要好好感谢一下哈佛的教学人员,整理出来一份很细致的方法对比说明
先上原图
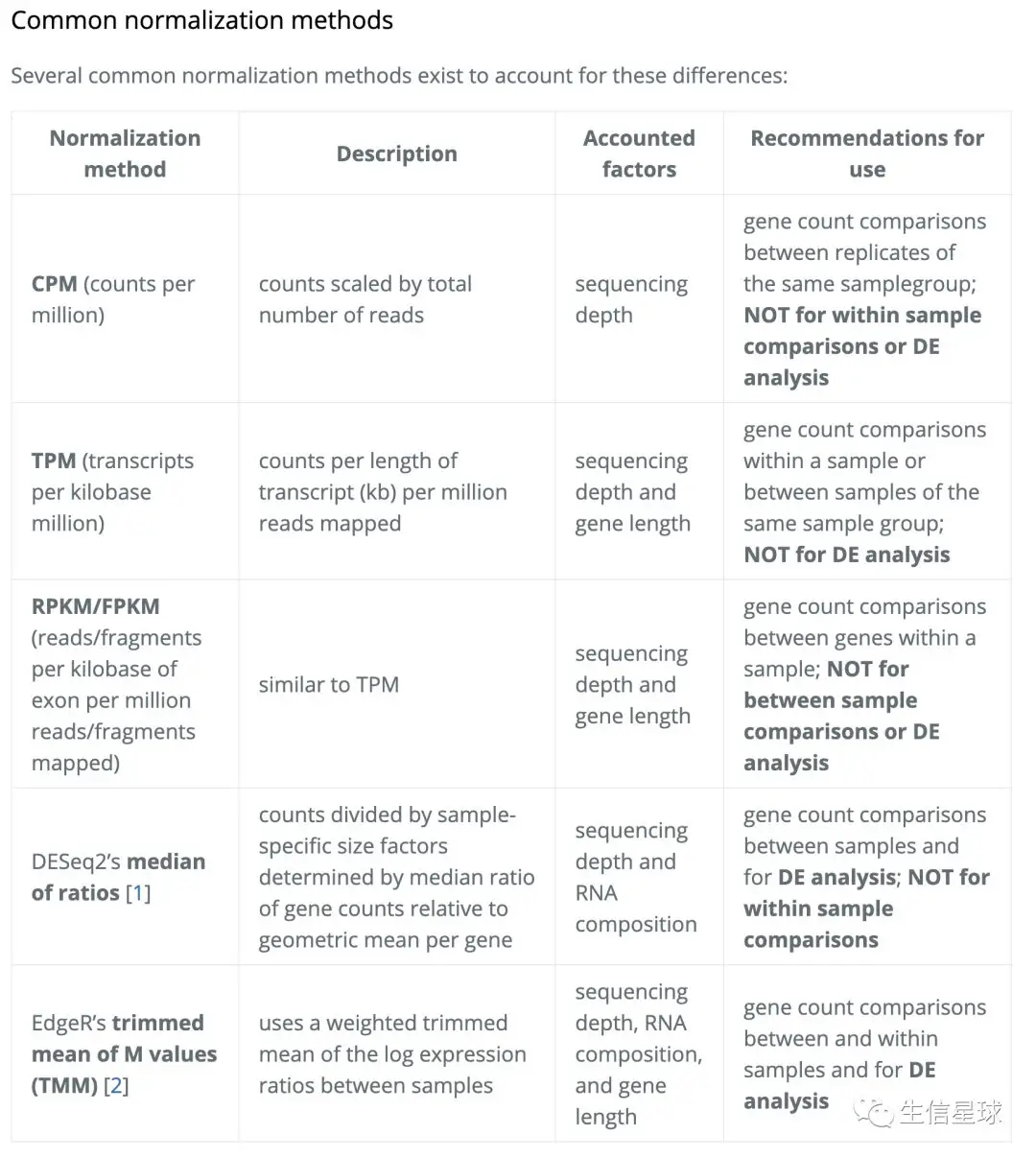
然后我再逐个解读
这些名词其实没必要翻译成中文,因为翻译过来可能更糊涂了。比如FPKM的翻译是:每1百万个map上的reads中map到外显子的每1K个碱基上的Fragments个数。而看它的计算方法其实最直观:
FPKM= read counts / (mapped reads (Millions) * exon length(KB))
CPM (counts per million)
- 主要适用于组内重复样本间比较
- **不适用于:**组间样本间比较(也就是差异分析);组内重复样本间比较
- 主要考虑的是:测序深度
TPM (transcripts per kilobase million)
- 主要适用于:组内重复样本间比较、组内样本内比较
- 不适用于:组间样本间比较(差异分析)
- 主要考虑的是:测序深度和基因长度
RPKM/FPKM
- 主要适用于:组内样本内比较
- 不适用于:组间样本间比较(差异分析)、组内重复样本间比较
- 主要考虑的是:测序深度和基因长度
这就很有意思了,为什么TPM和RPKM都是考虑了测序深度和基因长度,但RPKM却不能进行组内重复样本间比较呢?
这是因为RPKM或者RPKM算出来的标准化后的数值和TPM不同:每个样本中的TPM的总和都是一样的,而FPKM则不相等【先看这篇: 标准化进行时】:The total number of RPKM/FPKM normalized counts for each sample will be different.
总和不同当然不能直接进行比较了
DESeq2’s median of ratios
- 主要适用于:组内重复样本间比较、组间样本间比较(差异分析)
- 不适用于:组内样本内比较
- 主要考虑的是:测序深度和RNA组成
EdgeR’s trimmed mean of M values (TMM)
它应该是考虑问题最多的标准化方法
- 适用于:组内重复样本间比较、组间样本间比较(差异分析)、组内样本内比较
- 主要考虑的是:测序深度,基因长度和RNA组成
补充说明
下面是针对一些方法的补充
问题一:RPKM/FPKM
RPKM:以每个Read为一个单位,单端测序常用
FPKM:以Fragment一个单位,主要在双端测序
它们的差别是:FPKM在一对reads map上的情况下只计数1,而RPKM会计数2。因此为了准确,PE测序时我们认为一对reads比对上也只算一次,因此使用FPKM
一般无参转录组使用RSEM计算的结果,会给出FPKM和TPM两种结果
关于计算方法【难度在于准确计算基因长度】:看:「刘小泽RPKM概念及计算方法」https://www.jianshu.com/p/2bfa2b65a700
问题二:10X单细胞数据中的count需要计算TPM、FPKM等吗?
答案是不需要:https://kb.10xgenomics.com/hc/en-us/articles/115003684783-How-to-calculate-TPM-RPKM-or-FPKM-instead-of-counts-
计算TPM、FPKM的一个原因是:比较一个样本内的两个基因时,担心基因长度会造成影响。
- UMIs enable accurate quantitation of gene expression levels because we can tell when reads are generated from the same mRNA molecule.
- In traditional RNA-seq workflow, the probability of sampling a fragment from a long transcript is higher than from a short one.
- in 10x single cell 3’ or 5’ gene expression assay, this gene-length bias does not exist.
问题三:还有一种RPM和TPM一样吗?
它们的对比可以看:https://reneshbedre.github.io/blog/expression_units.html
- RPM:Reads per million mapped reads
- TPM:Transcript per million
- TPM proposed as an alternative to RPKM due to inaccuracy in RPKM measurement (Wagner et al., 2012)
RPM不考虑转录本长度,而TPM考虑;不过它和CPM倒是类似(分别是:Reads/Counts of exon model per Million mapped reads)
问题四:FPKM和TPM标准化过程的区别
- FPKM和RPKM:先按列(也就是这个样本的总count值)进行标准化,然后再对每个基因的长度进行标准化
- TPM:先对基因长度进行标准化,之后再对列(此时已经不再是原来count值的简单加和了)进行标准化
问题五:DEseq2的详细标准化方法
看哈佛的讲解,非常详细易懂:https://hbctraining.github.io/DGE_workshop/lessons/02_DGE_count_normalization.html
Step 1: creates a pseudo-reference sample (row-wise geometric mean)

Step 2: calculates ratio of each sample to the reference

Step 3: calculate the normalization factor for each sample (size factor)

Step 4: calculate the normalized count values using the normalization factor
问题六:如何计算TPM?
偶然看到一篇文章发在了Bioinformatics:https://github.com/ncbi/TPMCalculator
核心内容是:给他bam和GTF,给你算出来结果

另外,各种标准化计算的公式,都非常清楚了写在了:https://reneshbedre.github.io/blog/expression_units.html
可以自己根据公式去探索如何得到其中的每个数【当然好像也有一些R包可以做这个事情,但是需要时间去找、去验证】
其他的资源
视频:RPKM, FPKM and TPM, Clearly Explained!!! (https://www.youtube.com/watch?v=TTUrtCY2k-w)
文章:
浅谈RPKM,FPKM,RPM,TPM的区别 https://vip.biotrainee.com/d/63-rpkm-fpkm-rpm-tpm
CPM与TPM的区别:https://bioinformatics.stackexchange.com/questions/2298/difference-between-cpm-and-tpm-and-which-one-for-downstream-analysis