023-转录组谜团
刘小泽写于2018.8.17 通过一个小任务来帮助理解转录组测序的点滴
首先,设想手头有一个物种的三个基因ABC,它们的长度分别是10bp、100bp、1000bp,你想研究这个物种在两种不同环境下(野生型WT,热激发态HEAT)的差异缘由。 然后,你通过寻找其他文献发现,WT条件下,A基因的表达量要比B高两倍;另外你知道还有一个基因在两个不同条件下表达量是不同的,并且差异足够检测出来(但不知道是哪个基因)。其他基因在WT和HEAT中表达量一致。
这时,你采用了转录组测序进行比较WT和HEAT样本,但是每个样本只有一个重复。
不幸的是,你在做实验过程中出了点差错,发现得到的WT样本mRNA反转录得到的DNA量是HEAT的2倍,后来为了挽救,在上级测序前通过加测序标签将二者区分开来,然后将HEAT的DNA量也调整到了和WT一样
问题来了:
- 怎样从数据中看出你开始加了2倍的WT材料?
- 每个条件下的每个基因CPM值是什么意思?
- RPKM、TPM又是什么意思?
- 你怎么验证WT样本中A基因表达量是B基因的2倍?
- 怎么分辨在WT和HEAT中哪个基因产生了明显的差异?
- 如果要使CPM、RPKM、TPM得到一个比较合理的数字,那么应该测多少reads?
- 为了使它们看似合理,人工加上限制因子这种做法合适吗?
这些问题都会在后面进行解释
转录组测序前世今生
什么是转录组
转录组(transcriptome)广义上指某一生理条件下,细胞内所有转录产物的集合,包括信使RNA、核糖体RNA、转运RNA及非编码RNA;狭义上指所有mRNA的集合。它连接了基因组遗传信息与蛋白质组的生物功能。我们日常用的转录组测序一般是为了分析样本间基因表达量差异,当然还可以寻找可变剪切位点,发现新转录本等。
为什么有了转录组测序
测序技术的首次应用就是用来检测DNA分子的核酸组成的,但是后来人们除了组成成分以外,更想知道哪个成分有多少,这就是定量。测定基因的表达量有许多种方式,比如基因芯片、qPCR等。
基因芯片的开发使得一次性从一个基因组中获得大批基因表达量成为可能,转录组测序在芯片基础上,更加精准。芯片就像模拟信号,而转录组测序就是数字信号,他能检测到更多的差异表达基因(即动态范围大)。
什么是gene isoforms
isoforms翻译的话可以翻译成“亚型/异构体”,gene isoforms可以理解为一个基因的不同形态,就是由同一个基因座产生的mRNA,在转录起始位点(Transcription Start Sites, TSSs),编码蛋白序列(protein-coding DNA sequences, CDSs),非翻译区(Untranslated regions, UTRs)这些地方有差别,间接地改变了基因的功能。
一个基因座可以是一个基因,一个基因的一部分,或具有某种调控作用的DNA序列。基因座是染色体上的固定部位,在相同基因座上编码相同的DNA被称为等位基因。同一基因座上有相同的等位基因就是纯合子,相同基因座上是不同的等位基因就是杂合子。

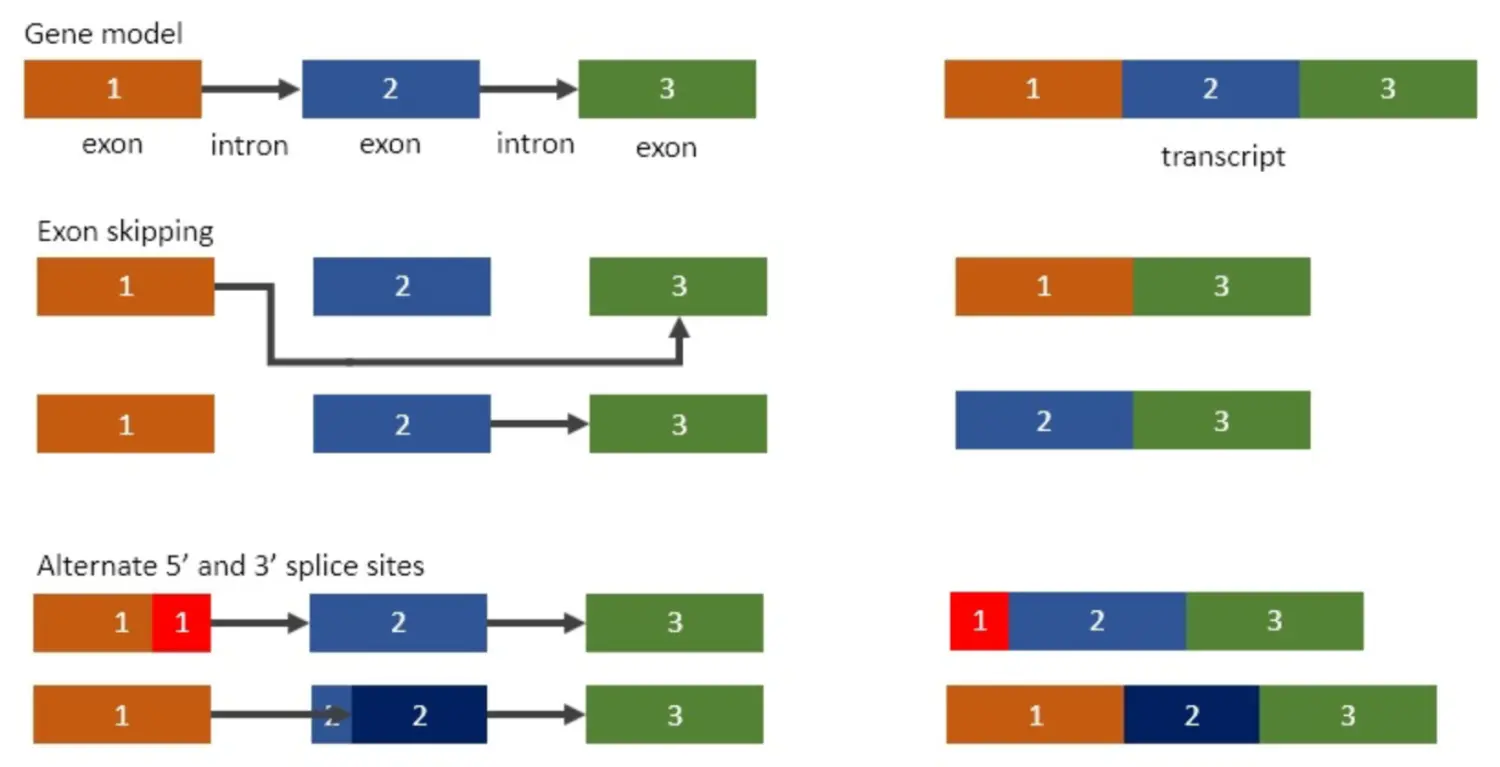
图中1、2、3是mRNA的三个外显子,由于连接方式不同,产生了三种isoforms
什么是可变剪切
它的学名是Alternative splicing, AS,又名选择性剪切。大多数真核基因转录产生的mRNA 前体一般按一种方式剪接产生出一种mRNA,结果只产生一种蛋白质。但有些基因产生的mRNA 前体可按不同的方式剪接,产生多于两种的mRNA。
首先看一下真核细胞基因的结构。真核生物基因序列包含了外显子exon、内含子intron,二者相互间隔。
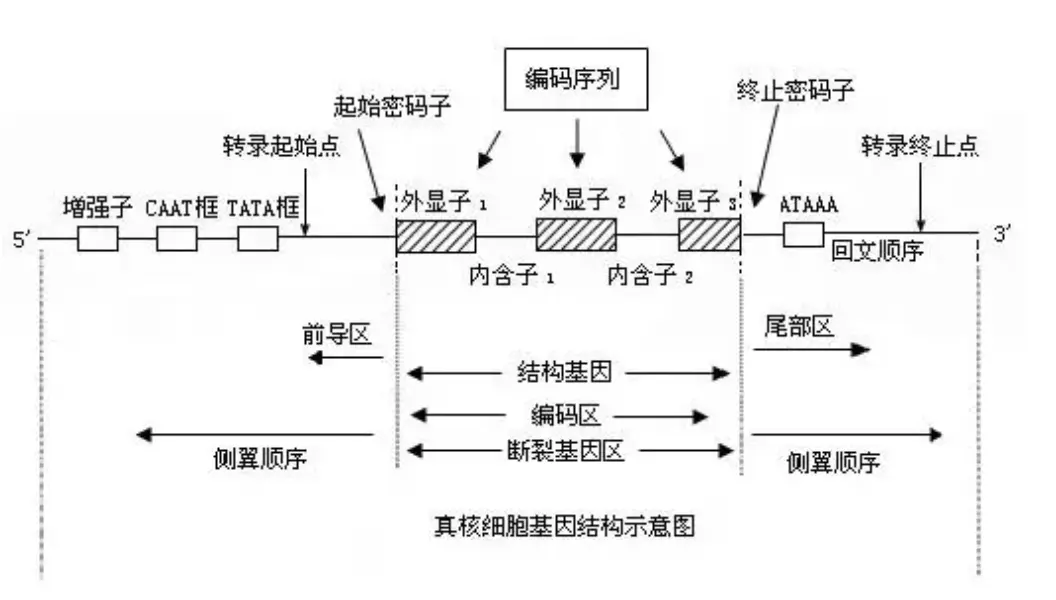
编码蛋白的成熟mRNA是mRNA前体经过剪切过的,外显子可以不按其线性次序剪接,内含子也可以不被切除而保留。因此成熟的mRNA中每一个外显子、内含子的存在与否都是不一定的。有5种类型:1、外显子跳跃(Exon skipping or cassette exon);2、内含子保留(Intron retention);3、5‘端可变剪切(Alternative donor 5’ site);4、3‘端可变剪切(Alternative acceptor 3’ site);5、特定外显子可变剪切(比如第一个或者最后一个外显子)(Mutually exclusive exons)
测序实质
RNA反转录成cDNA(cDNA, complementary DNA),测的就是cDNA,通过检测的cDNA表达量,可以推断出RNA的数量。看似流程很简单,就是数一数有多少DNA的片段,如果特定条件下,某个基因cDNA片段数量比较多,那么也就意味着原始RNA的含量也很高,即该基因表达量高。但是实际操作中,正是怎么计数,怎么比较才是分析的精髓。
分析的阻碍
- 测量标准:为了比较谁多谁少,一般都是采用相对定量的分析方法。但是同样的绝对数量对于不同的基数来讲,得到的相对值也是不同的。比如,第一次测A、B基因表达量是10,那么这一次中A基因的丰度就是10/(10+10)=50%;第二次测A、B、C表达量都是10,那么这一次,A的丰度就成了33.3%,但是能说第二次比第一次A基因的表达量丰度下降了吗?因此,只看表面的数字可能不能反映实际问题
- 测量方法:目前转录组测序采用的二代测序,还是测一段DNA的一小部分。由于可变剪切一般将内含子去除,而拼接不同的外显子,但毕竟它们“师出同门”,因此得到的转录本也是相似的,就像上面图中1、2组成的转录本a和1、3组成的转录本b都含有3,因此要将一小段比对到原始转录本就比较难(比如,一条reads符合3的一小部分,那么这条reads是属于转录本a还是转录本b呢?)
- 测量对象:mRNA不像DNA一样稳定,它很容易降解,因此它的丰度是时刻改变的。因此测转录组之前需要这个时间节点确保观察到了变化,并且这个变化与实验条件有关系。一般为了证明这一点,需要测定一个状态下的样本好几次,也就是所做的重复。差异基因的确定也必须通过重复来验证,也就是一个条件下几个重复得到的值如果都与标准条件下几个重复的值有差别,这才认为这个差异基因有效。
关于重复
推荐最少设置三个,五个更好。关于样本重复与测序深度的取舍,这一篇文章给出了解释:Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data
- 样本数的选取相比测序深度更能影响差异基因筛选的精确性;
- 表达量低的基因,reads数和样本数的选取对差异基因筛选有更大影响,而表达量高的基因,提高测序深度基本不会改变差异基因筛选的结果
总而言之,就寻找差异基因而言,还是建议多样本量;但是如果想研究可变剪切、发现新转录本的情况,还是要多测深度,加大reads数量
最好用的转录组流程?
看过这个就明白了:方案很多,几十上百个软件供你选择,其中好用的有很多,但是不会有最好的流程,只有自己搭配出适合自己的。与其选择流程,不如熟悉原理,在结果不合常理时知道怎么去纠正。转录组的基本流程用两套方法就能熟悉过来。

一般流程
一般就是:质控-》比对(alignment or mapping)-〉估算表达量(read counting)-》表达量比较(differential expression)。当然也有不需要比对就能进行量化分析的软件,比如kallisto【多说一句,它之所以可以跳过序列比对的步骤,是基于一个已经被论证的前提,即一条read具体比对到参考基因的什么位置上,并不影响最终的表达量结果。kallisto主要是确定一个 read 属于哪一个基因,而不关心这个 read 在基因上的位置】
比对环节有两个选择:一是比对参考基因组(genome),可以帮助发现新转录本以及gene isoforms;二是比对参考转录组(transcriptome),也就是在已知基因的前提下,更准确的定量样本中信息
有一种比对叫splice-aware
有许多测序reads是来自两个外显子的连接处(也就是剪切位点),如果要比对会参考基因组,reads的中间肯定会被加入一段空白(也就是原来的内含子)。相当于原来reads是脚踏两条船,现在两条船要回家,reads的腿就开始劈叉了。因此,对比软件必须要考虑到这一点,容许reads比对回去后,中间含有大大的空隙。
加入这种比对模式的软件有:
- Hisat2:与Bowtie2/Tophat2算法相似,但速度大提升
- Subjunc:专为基因表达分析打造
- BWA:它的mem算法支持剪切位点比对
- BBMap:支持非常长的剪切比对
- STAR:准确,提供counts定量数据
比对完如何定量?
最常用的三种进行相对定量的方法:
Counts:与转录本重叠的reads数
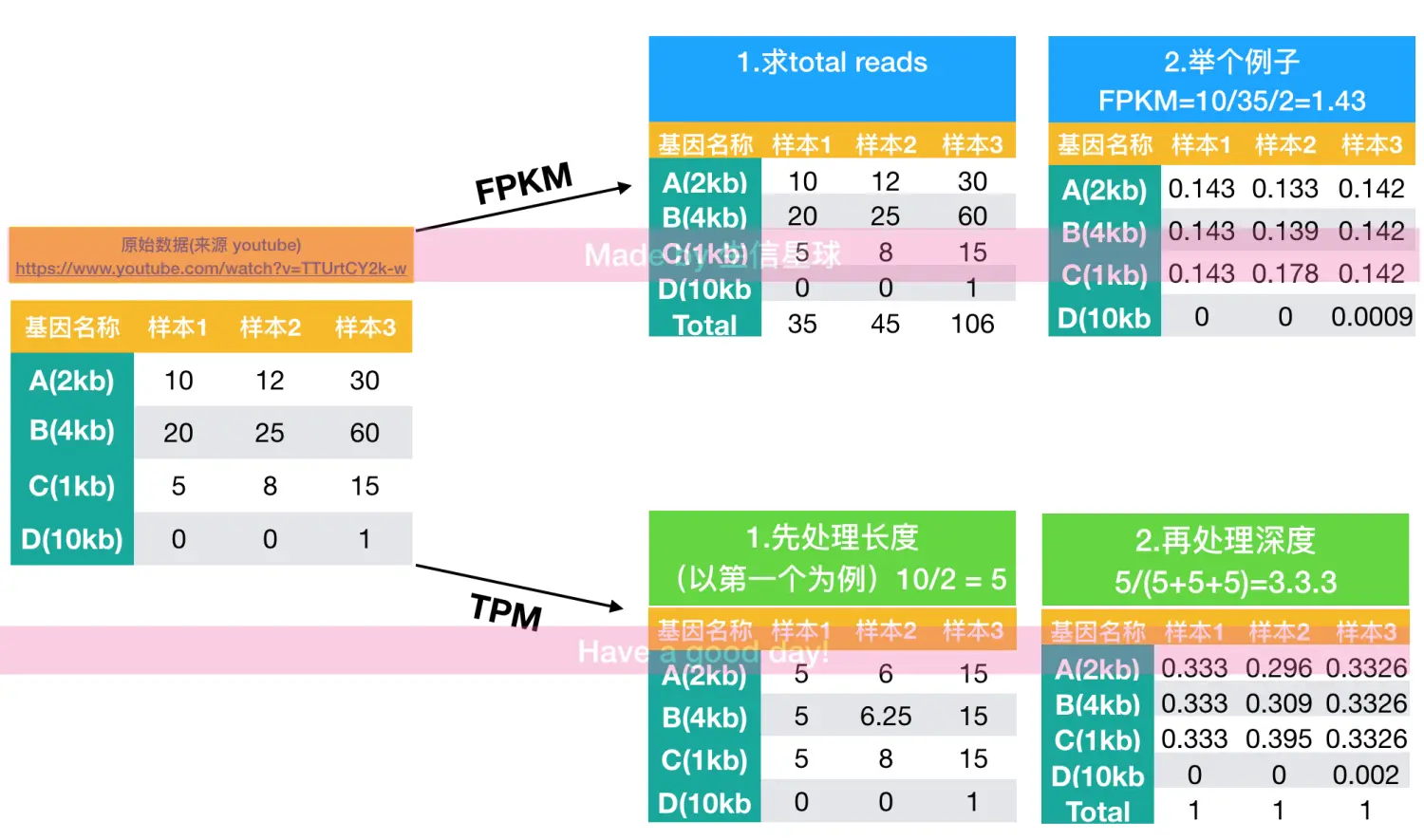
RPKM/FPKM:Reads/Fragments per kilobase of transcript per millions of read mapped【落在一个基因区域内的read counts数目取决于基因长度和测序深度,换句话说,一个基因越长,测序深度越高,落在其内部的reads数目就会相对越多。而为了比较不同样本中不同基因的表达量,就去除测序深度和基因长度的的影响。采用了两个标准化:reads数标准化和长度标准化】RPKM(A)=C/(N/10^6^*L/10^3^) ,其中C是唯一比对到转录本A的reads数,N是唯一比对到所有转录本的reads数,L是转录本A的长度。
建库测序是一个随机抽样的过程,而这个抽取的样品实际上是以 Fragments 为单位,而不是 Reads。因此,使用FPKM更为合理。当 single-end 测序的时候,RPKM 与 FPKM 是等价的;当 pair-end 测序的时候(一个fragment对应两条reads),应该使用 FPKM。
TPM: Transcripts per million reads。当样本差异过大,要强调准确度或者定量目标基因的表达量的时候,TPM是最有效的。TMP先处理基因长度问题,再处理测序深度
基因表达量的比较
- 样品内比较:比较同一个实验中基因表达量。例如,这个样本中A基因表达量高于B吗?
- 样品间比较:比较不同实验条件的样品。例如,不同实验条件下,A基因的表达量变化了吗?
一些名词
- sample:样本(包含了实验条件和重复),例如,我们说测10个样,也就是1个处理一个对照,各5个重复
- normalization:标准化,将各个样本的结果放在同一维度上,赋予它们可比性
- library size normalization:文库大小标准化,用来矫正不同实验条件下的测序深度(覆盖度)。例如,条件A相对B加了2倍的材料进行测序,这时就需要平衡两者
- effective length:有效长度,其实也就是长度越长的转录本,reads落在上面的数量越多,因此计算时需要平衡
- gene level:在基因层次上,将每个基因视作独立的转录本,而且均包含基因的所有外显子。但是还有一些情况需要更细致的分析,因此基因表达量需要建立在转录本层次上(transcript level)
- TMM(edegR) normalization: Trimmed mean of M values。edgeR软件使用的标准化算法叫TMM,它排除了一对实验条件下reads counts比率比较极端或者表达量的平均值比较极端的基因,从而估算的测序深度
- DESeq normalization:DEseq包使用的算法。它选出所有基因reads counts中位数的基因,估算它的测序深度