002-生信Linux文本处理三剑客
慢慢看,憋着急!很有用!
前言:
首先呢,在你的Linux系统中新建一个文件,Thanos.txt(紫薯侠赐予你力量)
# 敲入这一段,待会儿肯定给你解释清楚啥意思
echo -e "Hello world\nI love bioinformatics\n\nWelcome to bioinfoplanet\nAnd I will be happy here\n\nWelcome come to our Wechat public account\nbioinfoplanet" > Thanos.txt
然后你会发现多了一个紫薯侠文件,cat Thanos.txt 看是不是输出了几行字?真棒👍你已经开始了第一步
解释一下,echo是打印到终端,你可以理解为打印机,我们赋予它内容,他就给我们打印出来,到Thanos.txt这个本子上。
- -e : 内容中有特殊字符就需要加(比如上面👆的换行符\n)
- " “中就是要打印的内容
>输出到哪里
利用这个文件,你可以继续向下学习。当然如果你认为内容太少,你可以vi Thanos.txt 进入vi编辑器输入i编辑
文本查看:
查看命令
head, tail查看文件头尾-n查看指定行 (默认值为10行)cat将文件全部打出来less逐页显示文本-S规则输出-N显示行号less界面中可以移动光标,搜索关键字
less可以进行debug,查看中间输出结果 例如有一个管道程序sh1 test.txt | sh2 | sh3 > output.txt 【其中sh1-3都是命令】 在运行之前,我们可以看看每一步是否正确,这样使用: sh1 test.txt | less sh1 test.txt | sh2 | less sh1 test.txt | sh2 | sh3 | less
- 查看行数:wc -l 输出行数 (但是空行也会被算入)=> 解决:
grep -c "[^ \\n\\t]" file - 去除注释信息(元数据)后查看行数
一般基因组注释GTF文件开始都有几行的注释文件,需要去除以后再查看
grep -v '^@' *.gtf | wc -l这样做的好处就是接下来还能用awk直接处理列或者计算列数grep -v '^@' *.gtf | awk -F "\t" '{print NF};exit' - 查看列数【以tab分割为例】:
awk -F "\t" '{print NF ; exit}' *.bed
编辑器命令:
q:退出
g:第一行 G:最后一行
/
n向后匹配 N向前匹配
文件输出:
echo -n 不换行输出 -e 处理特殊字符
echo -e "nihao\nworld" echo -n "nihao\nworld"
=> nihao => nihao\nworld
=> world
>覆盖原文件 >>添加到原文件底部
正则表达式regular expression:
它的语法结构有两套系统组成,元字符(metacharacters) + 普通字符
那么问题来了:什么是元字符呢? A: 元字符是表达式的结构,骨架;比如,“我爱生信星球”这个句子中的主谓宾都是固定的,也就是它的元字符是固定的;普通字符可以不同,也就是会有各种语言版本的这句话,来表达我对生信星球的爱
元字符主要由以下字符组成:^ $ . [] {} - ? + () | \
| 表达式 | 描述 | 范例 |
|---|---|---|
| ^ | 行首标记 | ^bioinfoplanet 匹配以bioinfoplanet起始的行(以下简称bip) |
| $ | 行尾标记 | bip$匹配以bip结尾的行 |
| . | 任意字符 | b.p匹配任意代替.的字母,如bap,bbp…但不能代表两个如baap |
| [] | 其中任意一个 | bi[op]匹配bio或bip |
| [^] | 除了其中任一个 | bio[^pb]就是不能匹配biop & biob,其他任意 |
| [a-d] | 匹配指定范围内任一个 | 能匹配a-d任意一个字母 |
| {n} | 匹配之前n项 | [0-9]{2}匹配一个两位数[0-9][0-9] |
| {n, } | 至少匹配前面n次 | [0-9]{n, } 匹配至少是两位数的 |
| {n, m} | 最少匹配n次,最多m次 | [0-9]{2,4} 匹配两位数到四位数 |
| ? | 匹配之前1个或没有 | bi?p 匹配bip或bp |
| * | 匹配之前多个或没有 | bi*p匹配bp或bip/ biip/… |
| + | 匹配之前1个或多个 | bi+p匹配bip或biip/biiip/… |
| () | 匹配括号中的字符串 | (搭配?或 或+ 使用 )* bio(info)?匹配bio或bioinfo |
| | | 匹配两侧任一个 | bio\|info 匹配bio或info |
| \ | 转义 | bio\ +info 匹配bio+info,否则按+格式处理bioinfo |
()与[]的差别:()为多选,[]为单选, 对比下就知道
cat SRR519926_1.fastq | egrep 'TA(A+)TA' --color=always | head
@ 匹配了多种模式

cat SRR519926_1.fastq | egrep 'TA[A+]TA' --color=always | head
@ 仅匹配了一种模式

(e)grep:号称“Almost~最快文本搜索”
意思是 global search regular expression(RE) and print out the line
egrep 是grep的拓展模式,支持的元字符较多
生而为搜——它会在每一行搜索匹配内容
-c 显示有多少行被匹配到(count)
-v 过滤掉某些格式的行
-w 完全匹配【比如想删除匹配到abc的,不使用-w可能abc1和abcd都要被删除】
-o 只打印匹配到的内容
-i 忽略匹配字段和匹配内容的大小写
-A/B n: 输出匹配内容前/后 n行
--color=always/auto: 始终/自动高亮显示搜索字段
搜索特定信息
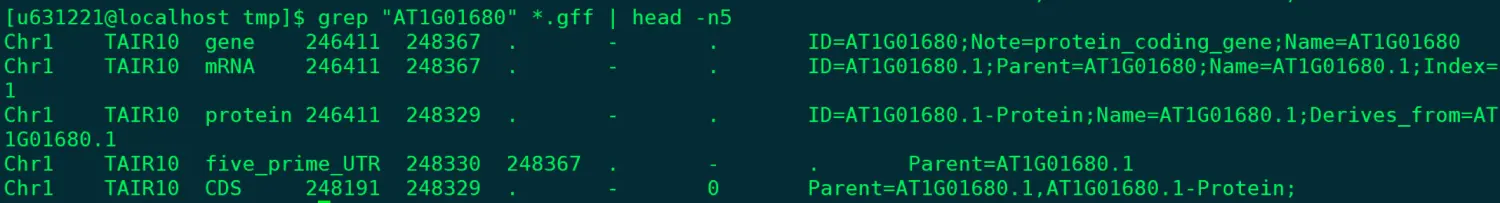
@ 搜索特定基因信息(在拟南芥gff文件中查找,下载地址见下方🌰) grep "AT1G01680" *.gff | head -n5
排除特定信息
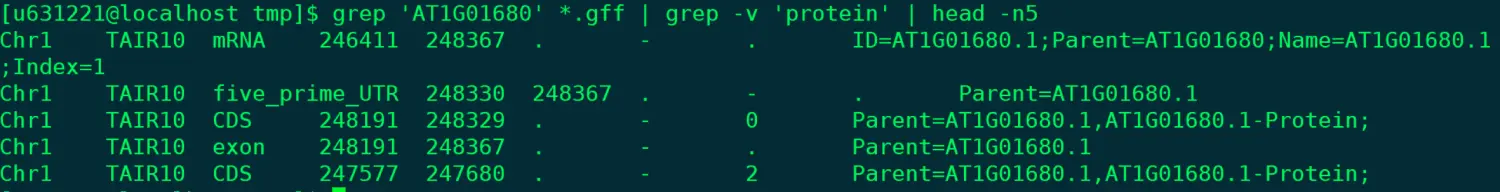
@ 还是搜索这个基因,但排除其中feature项的protein grep "AT1G01680" *.gff | grep -v "protein" | head -n5
查找某段序列并输出上下文
@ grep -A n显示后面n行; -B n显示前面n行 @ 下载基因组文件wget http://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_chromosome_files/TAIR10_chr_all.fas grep -A 2 "CAAATTGAATTAAG" *.fas => 得到下面👇的结果 => -B的用法你可以试一下

查找特定序列并计算出现了几次
grep -c "CAAATTGAATTAAG" *.fas 或者: grep "CAAATTGAATTAAG" *.fas | wc -l @ 如果单纯输出 就用grep -o精准匹配某个基因
@ 搭配正则表达式 @ 比如要匹配开头为AT1G250,结尾为3的基因名 grep "AT1G250.*3$" *.gff => 得到了两条AT1G25053 & AT1G25083

并非完美~
虽然grep很强大,但是他也并非十全十美,他的一个不足之处就在于,对于存在换行符的字符串,它会搜不到。例如我们想找TAIR10_chr_all.fas 中95-100之内的‘CCACT’ 碱基
先看一下
tail -n100 TAIR10_chr_all.fas | head -n5

用肉眼就能看到’CCACT’,但是如果用grep搜索 是没有结果的
tail -n100 TAIR10_chr_all.fas | head -n5 | egrep 'CCACT'
其实稍加改变,用grep还是能实现,就是不那么优雅:
tail -n100 TAIR10_chr_all.fas | head -n5 | tr -d '\n' | egrep 'CCACT'
tr -d(delete) 是删除特定字段
所以,有没有什么更快捷的办法呢? 有的~可以使用emboss套件下的dreg, 它是针对核酸;如果是氨基酸序列和翻译后的蛋白序列,使用preg
tail -n 1000 chr22.fa | head -n 5 | dreg -filter -pattern 'TAATA'
sed:
流编辑器~就像水流一样,按行从数据读取–执行命令–显示结果,一气呵成 整个过程在一个位于内存中叫Pattern Space模式空间中进行,因此不会改变原始文件内容
使用方法:
sed [options] 'Adress Command1;Command2;...' file...
[options]包括: -n: 只显示经过sed处理的行,保存在模式空间的未处理行不显示【常用】 -i:直接修改读取内容,不输出【慎用,他会修改源文件】 -r:使用拓展正则表达式
Adress工作范围: 可以指定为按行号工作或按过滤条件工作
按行号:使用单独的数字表示某一行;两个数字并逗号分隔表示范围;
n,m+表示从n向下m行;n~m表示从n开始的每m行比如我要输出偶数行: sed -n “2~2 p” Thanos.txt
按匹配条件:
'/pattern/command'在包括pattern的行中执行命令比如要打印包含bio的行:
sed -n '/bio/p' Thanos.txt比如打印包含bio与包含planet之间的所有行:sed -n '/bio/, /planet/p' Thanos.txt
还可以与行号连用:sed -n '/bio/, +6 p'Thanos.txt打印包含bio行以及下面6行
Command命令[多个命令用分号分隔]: p:复制模式空间的内容,一般与-n连用【否则会一次输出两次】 d:删除【按行号或匹配条件】
删除第5行后面所有的行
sed '5,$d'
i:插入
例如,要在文件开头插入一行,三列name、length、feature
sed '1i name\tlength\tfeature' Thanos.txt
a:追加
例如,要在末尾追加内容【$表示最后一行】
sed '$a bioinfoplanet' Thanos.txt
c:替换【与a用法相似】 n:匹配的行向下移一行进行操作
例如:
seq 6 | sed '2{n;d}'输出的就是除了3以外的数字 再如:seq 6 |sed 'n;d'输出奇数seq 6 | sed -n 'n;p'输出偶数
!:反向执行
sed '/bio/!d' Thanos.txt效果等于sed -n '/bio/p' Thanos.txt
=:打印行号
sed '/bio/!d;=' Thanos.txt
s:替换【支持正则】
s/pattern/replace/flags
其中pattern是支持正则的 flags包括:n(替换第n个匹配);g(全局匹配);p(与-n搭配,输出修改的行);i(忽略大小写);w(保存修改的行到一个新文件)
一种特殊情况,还比较常用,就是如果替换
/怎么办? 方法一:使用转义符\/方法二 :用@ | ! ^替换 例如:要更改当前目录中的部分内容pwd | sed 's@/home/tmp/bio@/home/tmp/bioinfo@'
还有一种特殊情况,就是有时候不想替换掉,只是想把pattern的这部分内容与replace的内容一起输出来:
sed 's/[[:upper:]]/ word = &/' Thanos.txt意思就是:将Thanos.txt中的大写字母放到&的位置,输出格式就是:word = 大写字母。【特殊字符&用来存储pattern中的内容】
实用的sed单行命令:
@ 删除空行
sed '/^$/d'
@ 每一行下增加一行空行
sed G
@ 每三行增加一行空白行
sed '0~3G'
@ 在匹配的pattern后面加入一行空白行
sed '/pattern/G'
@ cat的功能实现
sed ''
@ head的功能实现
sed '3 q' =》输出前三行
@ tee功能实现
sed -n 'p; w newfile'
@ grep功能实现
sed -n '/pattern/p'
@ grep -v功能实现
sed -n '/pattern/p!'
@ 计算行数
sed -n '$='
@ 多个内容同时替换(例如将1、2、3替换成4)
sed 's/1\|2\|3/4/'
@ 显示包含“haha”、“xixi”、“yeah”的行
sed '/haha/d!; /xixi/d!; /yeah/d!'
awk:号称“Almost~最强文本操作”
“awk w(o/a)rd”—— 让看似繁复无章的文字遇见它就秒变尴尬😅
**工作职责:**主打行【如果一个文本文档是一张表格,每一行代表一个记录,每一列代表域,awk就是处理记录专用】
工作流程: 先逐行读取并记录,将行信息整合入$0 => 指定分隔符(默认空格),分割成各个列$1,$2,$3...;
再执行awk 'pattern1 {command1}; pattern2 {command2}...'
⚠️:情况一:如果缺失pattern【也就是利用下面几种运算进行的匹配】,直接执行{command}; 情况二:缺失{command}【在{}中输入的命令,如print】,执行pattern
现在不太懂也没关系,通过实例练习你就明白,下面的实例我会描述对应哪种情况
模拟cat打印行
awk '{print $0}' Thanos.txt | head -n3 @ 【因为没有pattern,所以直接执行打印命令】~属于情况一选择列打印
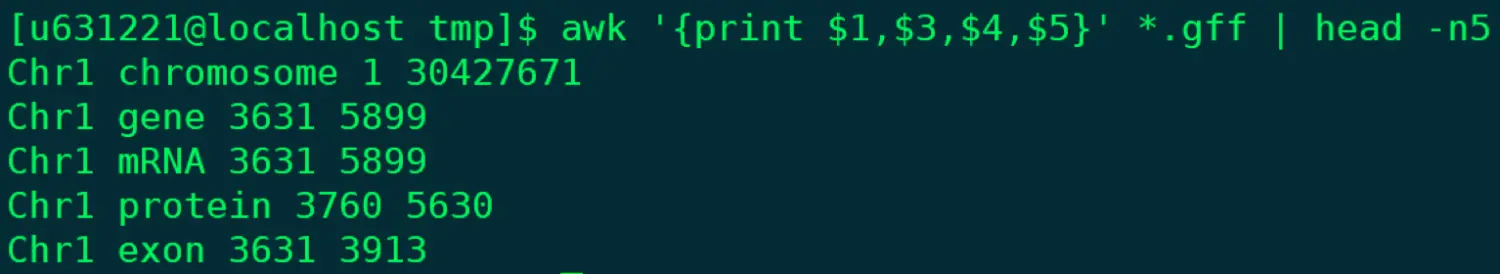
@ 利用.gff文件练习 ~属于情况一 awk '{print $1,$3,$4,$5}' *.gff | head -n5 【当然,这里默认是空格分割,如果想tab分割呢?】 awk '{[print $1 "\t" $3 "\t" $4 "\t" $5 "\t"]}' @ 一会练习完下面的cut命令,看看有什么发现?
列重排
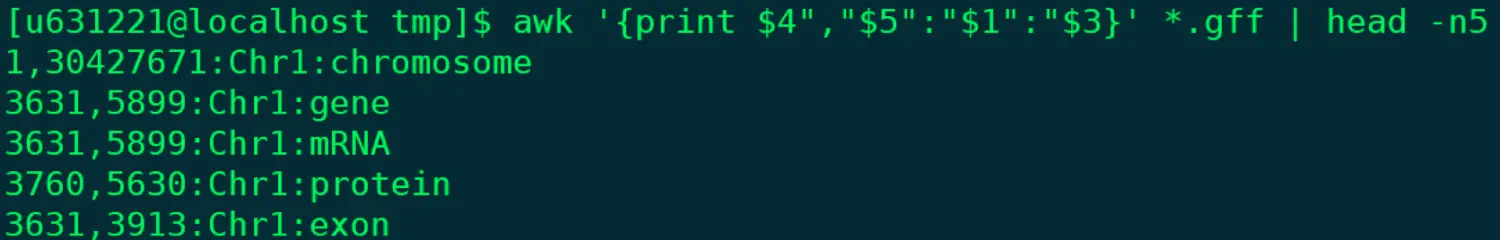
@ 这个功能相当于下面cut的升级版 ~ 属于情况一 @ 可以改变列的顺序,并且可以自定义分隔符 awk '{print $4","$5":"$1":"$3}' *.gff | head -n5 @ 随心所欲,只为你改变!
转换格式
比如将gff/gtf格式转为bed格式 【注意两点:一、转换后的bed格式分隔符为tab; 二、bed与gff的坐标格式不同:0-坐标系统有BAM、BED、BCFv2、PSL;1-坐标系统有SAM、VCF、GFF、wiggle。需要做出调整】
cat test.gtf | awk '{print $1 "\t" $4-1 "\t" $5}' > test.bed如果单有这些简单的功能,还算不上Almost最强,加上下面这些你再试试?
刘小泽喊你进阶啦!
逻辑运算(<, > , <=, >=, ==, !=)
数学运算(+,-, *, /, %)
关系运算(与&&, 或||, 非!)
正则运算 (实则就是将你想匹配的东西放在
/ /里,然后在它前面加~匹配,!~不匹配)举个小例子1:
@ 我想匹配在3号染色体上,长度大于1.5k的注释,看前五行 @ ~情况二,我们只需要pattern就好 awk '$5-$4 > 15000 && $1 ~/Chr3/' *.gff | head -n5
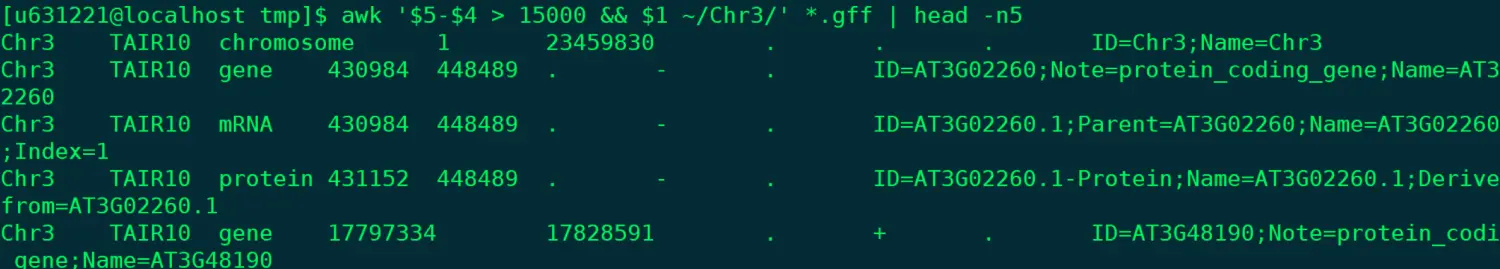
> 然后自己再试试匹配1号或2号染色体上,长度小于1.5k的注释,默认输出前10行
- 小例子2:
```平均值
@ 我想看一下3号染色体上编码区(CDS)的平均值 ~情况二
awk 'BEGIN {len=0;line=0}; $1 ~/Chr3/ && $3 ~/CDS/ { len += ( $5 - $4 );line += 1}; END {print "CDS_mean=" len/line}' *.gff
=>
CDS_mean=225.019
【这里 $1 ~/Chr3/ 中的~是为了匹配正则表达式】
@ 那么你试试看,1号染色体上外显子exon的平均长度吧
@ 此处只是用来练习命令,并不表示真实长度。因为重叠的区域(比如起始位点一样,终止位点不同)这样会被记录两次,结果是不准确的;如果要精确统计,需要用编程去重以后来实现
```
特殊的变量:
(上面说的)$ Num表示哪一个字块(这里再次提醒:$0代表当前内容)
NR代表当前所在行号,想要打印3-4行内容:
head -4|tail -2, 就等同于awk ‘NR>=3 && NR<=4 {print $0}‘`NF表示目前的记录被分割的字段的数目,即Number of Field
例如要输出文件的列数【默认空格分割,这里设为tab分割】: awk -F “\t” ‘{print NF; exit}’ test.bed
FS 指定列分隔符
OFS 指定列输出分隔符
例如将上游文件的默认分隔符\t更换成, 并输出3-5列
cat test.txt | awk '{FS="\t"; OFS=",";}{print $3,$4,$5}'
常用的还有:
cut:提取
-f 提取指定字段(filed)
cut -f 1,3,4,5 GFF3_genes.gff | head也可以cut -f 1-5-d: 指定分隔符(默认
\t)-c 截取一定范围的字符,例如
cut -c1-3就是截取三个字符例如,将bed文件第一列的染色体编号去掉 awk ‘{print $1}’ *.bed | cut -c4 反之,要再添加上的话:
awk '{print $1}' *.bed | cut -c4 | awk '{print "chr"$1}'
uniq: 去重
与sort连用,-c 在每列旁边显示该行重复出现的次数 -d : 只输出重复的行
column:格式化输出
一般cut后的结果参差不齐,可以用它对齐输出结果,默认\t
指定分隔符:以逗号为例, -t -s ','
【⚠️:他的使用只是为了好看,不要把它column处理的结果交给下游继续处理,这样会让文本解析速度下降。😋看来Linux还是更习惯参差不齐的文本】
sort: 排序
- -k 被排序的部位,包含
start和stop两部分, 二者,隔开;每次使用都要加上这两部分【默认排序第一列】 - -n 按数字大小排序
sort -k1,1 -k2,2n my.bed对第一列首字母按字符排序,对第二列按数值 - -r 降序(默认升序)【与-k连用表示对某一列逆序】
- -t 指定字块分隔符(默认tab)
- -c 检查是否按照某种方式排过顺序【
echo $?返回0表示执行成功】 - -V 排序时不用ASCII码方式排列,就显示为正常的排序方式 【比如三个染色体编号chr1, chr2, chr11。不加-V排序结果就是chr1– chr11 – chr2; 加了-V就是:chr1 – chr2 – chr11】 【一般数据处理过程中不用这个操作,原始的格式处理更快】
join:连接
要求两个文件之间必须有共同点,所以使用join前必须先将文件排序
格式:join -1 <file1_field> -2 <file2_field> <file1> <file2>
共同点通过-1 、 -2传递进来,比如说两个文件的某一列有共同点
例如一个bed文件前三列如下:my.bed
chr1 34 36
chr2 38 40
chr1 25 39
chr3 12 18
一个染色体长度文件为:length.txt
chr1 54362
chr2 35613
chr3 46612
join这两个文件之前先sort
sort -k1,1 my.bed > sorted.bedsort -c -k1,1 length.txt以双方第一列为共同点拼接:join -1 1 -2 1 sorted.bed length.txt > with_length.txt
【假如两个文件没有共同点,比如length.txt少了chr2数据,那么join后的文件也缺少chr2数据】
-a 选项指定哪个文件可以不遵循,在没有共同点时可以单列出来
举个🌰【很大的那种!】:
首先输出一下未排序文件,你可以自己下一个小数据(43M)试试, 人类的比较大(1.2G)我就不下了
wget http://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes.gff【这是拟南芥的基因注释文件,当练手很好用】
先练习下之前学的一些处理:
@0 拿到手数据,可以先ll -h *.gff 看一下文件大小 less -SN *.gff 看一下文本内容 @1 去除多余的#注释行 与 空行, 打印行号 grep -v "#" *.gff | grep -v "^$" | wc -l => 590264行(一般这种使用量大的数据是没有多余行的,但是人类的有。这里只是为了演示用法) @2 截取gff文件的1-5列,将第二列除去, 输出默认的前10行到test.txt中 cut -f 1,3,4,5 *.gff > test.txt cat test.txt | head => Chr1 chromosome 1 30427671 Chr1 gene 3631 5899 Chr1 mRNA 3631 5899 Chr1 protein 3760 5630 Chr1 exon 3631 3913 Chr1 five_prime_UTR 3631 3759 Chr1 CDS 3760 3913 Chr1 exon 3996 4276 Chr1 CDS 3996 4276 Chr1 exon 4486 4605
接下来对截取的test.txt进行处理
@3 想根据第二列的feature进行排序【注意sort -k的使用!】 sort -k 2,2 test.txt | head -n5 => Chr1 CDS 3760 3913 Chr1 CDS 3996 4276 Chr1 chromosome 1 30427671 Chr1 exon 3631 3913 Chr1 exon 3996 4276 @4 【进阶】先根据第一列Chr数字大小降序排序,再根据第二列排序 你是不是试过了这个》sort -k 1,1nr -k 2,2 *.txt | head -n5 你会发现输出的结果第一列还是Chr1开头,并没有降序,为什么呢? =》原因就是,-k 1,1还是根据第一个字段的全部排序,还是根据Chr1的‘C’进行匹配,其实我们只想用第一个字段的数字(也就是第一个字段的第四个字符)进行匹配 =》如何实现?其实sort -k参数 内置了这个功能。使用-k 1.4,1.4 就是根据这种特定方式匹配啦 sort -k 1.4,1.4nr -k 2,2 *.txt | head -n5 => Chr5 CDS 10001590 10001736 Chr5 CDS 10004720 10004824 Chr5 CDS 10004720 10004824 Chr5 CDS 10005070 10005255 Chr5 CDS 10005070 10005255 有没有很好用?!
如果只是想统计一下整体的feature(第三列)情况,可以这样:
cut -f 3 *.gff |sort|uniq -c >feature.txt => 197160 CDS 7 chromosome 215909 exon 34621 five_prime_UTR 28775 gene 35386 mRNA 3911 mRNA_TE_gene 180 miRNA 480 ncRNA 35386 protein 924 pseudogene 1274 pseudogenic_exon 926 pseudogenic_transcript 15 rRNA 13 snRNA 71 snoRNA 689 tRNA 30634 three_prime_UTR 3903 transposable_element_gene想看哪个feature最多?没问题,一步搞定!
sort -k 1r feature.txt => 215909 exon 197160 CDS 35386 protein 35386 mRNA 34621 five_prime_UTR 30634 three_prime_UTR 28775 gene 3911 mRNA_TE_gene 3903 transposable_element_gene 1274 pseudogenic_exon 926 pseudogenic_transcript 924 pseudogene 689 tRNA 480 ncRNA 180 miRNA 71 snoRNA 15 rRNA 13 snRNA 7 chromosome**【小练习1:】**GENCODE下载人类基因组GRCh38注释gff3 ,正好练习下数据库使用
然后统计人类的基因组 feature想偷懒?给你个机会~ wget -c ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_28/gencode.v28.annotation.gff3.gz
应该得到如下结果:=>
1237914 exon
735618 CDS
203835 transcript
148007 five_prime_UTR
144591 three_prime_UTR
85439 start_codon
77451 stop_codon
58381 gene
119 stop_codon_redefined_as_selenocysteine
【小练习2:】 相信你能够完成上面的内容,输出了结果。
**小前言:**我们知道,Linux基于Unix开发,肯定要继承unix的精华,那就是利用小程序的整合去完成大任务。Linux中的管道命令
|就是这样一种体现。管道中的数据可以不被写入磁盘,在更高速的内存中进行处理,就像空中绿道,不被道路上拥挤的车流阻拦,你可以在空中自由骑行,肯定效率高很多,心情也更舒畅~**问题来了:**你能否用一行管道命令从读取文件开始到输出排序好的feature文件呢?
提示:读取|截取|排序|统计|统计后排序