204-bedtools的简单操作版本
刘小泽写于2020.8.14
前言
之前初识bedtools的时候根据官网教程写了一个接近于实战的教程: 2019 和豆豆一起跟着官网学习 bedtools
但是,如果要想快速上手操作的话,可以使用更简单的数据
1 bedtools intersect
内容依然来自官网:https://bedtools.readthedocs.io/en/latest/content/tools/intersect.html
单个文件的操作是:
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
$ bedtools intersect -a A.bed -b B.bed
chr1 15 20

多个文件的操作是:

数据准备
$ cat query.bed
chr1 1 20
chr1 40 45
chr1 70 90
chr1 105 120
chr2 1 20
chr2 40 45
chr2 70 90
chr2 105 120
chr3 1 20
chr3 40 45
chr3 70 90
chr3 105 120
chr3 150 200
chr4 10 20
$ cat d1.bed
chr1 5 25
chr1 65 75
chr1 95 100
chr2 5 25
chr2 65 75
chr2 95 100
chr3 5 25
chr3 65 75
chr3 95 100
$ cat d2.bed
chr1 40 50
chr1 110 125
chr2 40 50
chr2 110 125
chr3 40 50
chr3 110 125
$ cat d3.bed
chr1 85 115
chr2 85 115
chr3 85 115
最简单的操作
$ bedtools intersect -a query.bed \
-b d1.bed d2.bed d3.bed
chr1 5 20
chr1 40 45
chr1 70 75
chr1 85 90
chr1 110 120
chr1 105 115
chr2 5 20
chr2 40 45
chr2 70 75
chr2 85 90
chr2 110 120
chr2 105 115
chr3 5 20
chr3 40 45
chr3 70 75
chr3 85 90
chr3 110 120
chr3 105 115
但通过这个最终结果,我们不知道是query.bed和哪个文件产生的交集
看更详细的结果报告 =》 -wa -wb
加入-wa,-wb参数,分别可以输出query文件的区间以及target文件的区间
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted
chr1 1 20 1 chr1 5 25
chr1 40 45 2 chr1 40 50
chr1 70 90 1 chr1 65 75
chr1 70 90 3 chr1 85 115
chr1 105 120 2 chr1 110 125
chr1 105 120 3 chr1 85 115
chr2 1 20 1 chr2 5 25
chr2 40 45 2 chr2 40 50
chr2 70 90 1 chr2 65 75
chr2 70 90 3 chr2 85 115
chr2 105 120 2 chr2 110 125
chr2 105 120 3 chr2 85 115
chr3 1 20 1 chr3 5 25
chr3 40 45 2 chr3 40 50
chr3 70 90 1 chr3 65 75
chr3 70 90 3 chr3 85 115
chr3 105 120 2 chr3 110 125
chr3 105 120 3 chr3 85 115
现在看到中间部分的1、2、3,它们实际上指的是target文件的位置,不过这样还是需要我们自己去做对应
让中间的文件名称更清楚 =》 -names
使用-names对各个target文件指定一个名称
-sorted的含义是:对染色体和起始坐标进行排序,而且文件越大,sort的优势越明显
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-names d1 d2 d3 \
-sorted
chr1 1 20 d1 chr1 5 25
chr1 40 45 d2 chr1 40 50
chr1 70 90 d1 chr1 65 75
chr1 70 90 d3 chr1 85 115
chr1 105 120 d2 chr1 110 125
chr1 105 120 d3 chr1 85 115
chr2 1 20 d1 chr2 5 25
chr2 40 45 d2 chr2 40 50
chr2 70 90 d1 chr2 65 75
chr2 70 90 d3 chr2 85 115
chr2 105 120 d2 chr2 110 125
chr2 105 120 d3 chr2 85 115
chr3 1 20 d1 chr3 5 25
chr3 40 45 d2 chr3 40 50
chr3 70 90 d1 chr3 65 75
chr3 70 90 d3 chr3 85 115
chr3 105 120 d2 chr3 110 125
chr3 105 120 d3 chr3 85 115
还可以直接把对应的target文件名放在中间 =》 -filenames
bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted \
-filenames
chr1 1 20 d1.bed chr1 5 25
chr1 40 45 d2.bed chr1 40 50
chr1 70 90 d1.bed chr1 65 75
chr1 70 90 d3.bed chr1 85 115
chr1 105 120 d2.bed chr1 110 125
chr1 105 120 d3.bed chr1 85 115
chr2 1 20 d1.bed chr2 5 25
chr2 40 45 d2.bed chr2 40 50
chr2 70 90 d1.bed chr2 65 75
chr2 70 90 d3.bed chr2 85 115
chr2 105 120 d2.bed chr2 110 125
chr2 105 120 d3.bed chr2 85 115
chr3 1 20 d1.bed chr3 5 25
chr3 40 45 d2.bed chr3 40 50
chr3 70 90 d1.bed chr3 65 75
chr3 70 90 d3.bed chr3 85 115
chr3 105 120 d2.bed chr3 110 125
chr3 105 120 d3.bed chr3 85 115
取出没有任何交集的部分 =》 -v参数
$ bedtools intersect -wa -wb \
-a query.bed \
-b d1.bed d2.bed d3.bed \
-sorted \
-names d1 d2 d3
-f 1.0
chr1 40 45 d2 chr1 40 50
chr2 40 45 d2 chr2 40 50
chr3 40 45 d2 chr3 40 50
只输出交集的bp数量 =》 -wo(Write the amount of overlap bp)
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ bedtools intersect -a A.bed -b B.bed -wo
# 最后一列表示5bp 和 2bp
chr1 10 20 chr1 15 20 5
chr1 10 20 chr1 18 25 2
不管有没有交集,都输出bp数量 =》 -wao
# 没有交集的输出结果就是0,比如最后一行
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ bedtools intersect -a A.bed -b B.bed -wao
chr1 10 20 chr1 15 20 5
chr1 10 20 chr1 18 25 2
chr1 30 40 . -1 -1 0
如果只想知道A文件的哪一行在B文件有交集,而不关心交集是啥 =》-u(unique)
# 如果不带u,就输出所有的交集
$ cat A.bed
chr1 10 20
$ cat B.bed
chr1 15 20
chr1 17 22
$ bedtools intersect -a A.bed -b B.bed
chr1 15 20
chr1 17 20
# 带上u,就只输出哪一行产生了交集
$ cat A.bed
chr1 10 20
$ cat B.bed
chr1 15 20
chr1 17 22
$ bedtools intersect -a A.bed -b B.bed -u
chr1 10 20
如果只想知道A文件每一行在B文件中有多少交集 =》 -C
$ cat A.bed
chr1 10 20
chr1 30 40
$ cat B.bed
chr1 15 20
chr1 18 25
$ bedtools intersect -a A.bed -b B.bed -c
chr1 10 20 2
chr1 30 40 0
默认交集是1bp,如果想修改 =》 -f(overlap fraction)
比如要求至少存在50%的交集才算
$ cat A.bed
chr1 100 200
$ cat B.bed
chr1 130 201
chr1 180 220
$ bedtools intersect -a A.bed -b B.bed -f 0.50 -wa -wb
chr1 100 200 chr1 130 201
如果存在链的信息,可以强制在同一条链寻找交集 =》 -s
$ cat A.bed
chr1 100 200 a1 100 +
$ cat B.bed
chr1 130 201 b1 100 -
chr1 132 203 b2 100 +
$ bedtools intersect -a A.bed -b B.bed -wa -wb -s
chr1 100 200 a1 100 + chr1 132 203 b2 100 +
如果存在链的信息,可以强制在不同链寻找交集 =》 -S
$ cat A.bed
chr1 100 200 a1 100 +
$ cat B.bed
chr1 130 201 b1 100 -
chr1 132 203 b2 100 +
$ bedtools intersect -a A.bed -b B.bed -wa -wb -S
chr1 100 200 a1 100 + chr1 130 201 b1 100 -
取bam和一个bed坐标文件的交集 =》 -abam
默认是输出bam结果
$ bedtools intersect -abam reads.unsorted.bam -b simreps.bed | \
samtools view - | \
head -2
BERTHA_0001:3:1:15:1362#0 99 chr4 9236904 0 50M = 9242033 5 1 7 9
AGACGTTAACTTTACACACCTCTGCCAAGGTCCTCATCCTTGTATTGAAG W c T U ] b \ g c e g X g f c b f c c b d d g g V Y P W W _
\c`dcdabdfW^a^gggfgd XT:A:R NM:i:0 SM:i:0 AM:i:0 X0:i:19 X1:i:2 XM:i:0 XO:i:0 XG:i:0 MD:Z:50
BERTHA _0001:3:1:16:994#0 83 chr6 114221672 37 25S6M1I11M7S =
114216196 -5493 G A A A G G C C A G A G T A T A G A A T A A A C A C A A C A A T G T C C A A G G T A C A C T G T T A
gffeaaddddggggggedgcgeggdegggggffcgggggggegdfggfgf XT:A:M NM:i:3 SM:i:37 AM:i:37 XM:i:2 X O : i :
1 XG:i:1 MD:Z:6A6T3
取bam和一个bed坐标文件的交集,并且输出bed结果 =》-abam + -bed
$ bedtools intersect -abam reads.unsorted.bam -b simreps.bed -bed | head -10
chr4 9236903 9236953 BERTHA_0001:3:1:15:1362#0/1 0 +
chr6 114221671 114221721 BERTHA_0001:3:1:16:994#0/1 37 -
chr8 43835329 43835379 BERTHA_0001:3:1:16:594#0/2 0 -
chr4 49110668 49110718 BERTHA_0001:3:1:31:487#0/1 23 +
chr19 27732052 27732102 BERTHA_0001:3:1:32:890#0/2 46 +
chr19 27732012 27732062 BERTHA_0001:3:1:45:1135#0/1 37 +
chr10 117494252 117494302 BERTHA_0001:3:1:68:627#0/1 37 -
chr19 27731966 27732016 BERTHA_0001:3:1:83:931#0/2 9 +
chr8 48660075 48660125 BERTHA_0001:3:1:86:608#0/2 37 -
chr9 34986400 34986450 BERTHA_0001:3:1:113:183#0/2 37 -
有时需要重新对染色体坐标排序,并重新指定 =》-g
比如原来的genome大小文件是:
$ cat hg19.genome
chr1 249250621
chr10 135534747
chr11 135006516
chr12 133851895
chr13 115169878
chr14 107349540
chr15 102531392
chr16 90354753
chr17 81195210
chr18 78077248
chr19 59128983
chr2 243199373
chr20 63025520
chr21 48129895
chr22 51304566
chr3 198022430
chr4 191154276
chr5 180915260
chr6 171115067
chr7 159138663
chr8 146364022
chr9 141213431
chrM 16571
chrX 155270560
chrY 59373566
现在需要按数值升序排序,而不是按照ASCII排序
$ sort -k1,1V hg19.genome > hg19.versionsorted.genome
$ cat hg19.versionsorted.genome
chr1 249250621
chr2 243199373
chr3 198022430
chr4 191154276
chr5 180915260
chr6 171115067
chr7 159138663
chr8 146364022
chr9 141213431
chr10 135534747
chr11 135006516
chr12 133851895
chr13 115169878
chr14 107349540
chr15 102531392
chr16 90354753
chr17 81195210
chr18 78077248
chr19 59128983
chr20 63025520
chr21 48129895
chr22 51304566
chrM 16571
chrX 155270560
chrY 59373566
最后再按排序后的genome文件找交集
$ bedtools intersect -a a.versionsorted.bam -b b.versionsorted.bed \
-sorted \
-g hg19.versionsorted.genome
2 有趣的小功能
只列出最简单的操作,如果遇到类似的问题知道用什么命令即可 还有很多参数就不一一列举,到时再查 全部功能如下:

2.1 从fasta中提取指定坐标的序列 =》getfasta
-fi指定fasta文件;-bed指定区间坐标
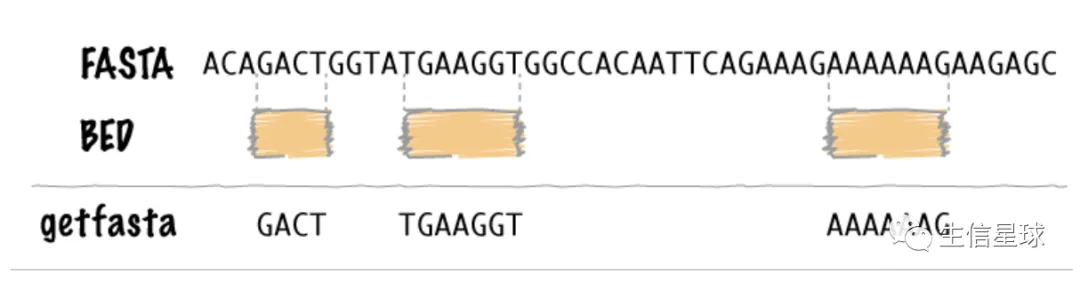
$ cat test.fa
>chr1
AAAAAAAACCCCCCCCCCCCCGCTACTGGGGGGGGGGGGGGGGGG
$ cat test.bed
chr1 5 10
$ bedtools getfasta -fi test.fa -bed test.bed
>chr1:5-10
AAACC
# 指定输出文件
$ bedtools getfasta -fi test.fa -bed test.bed -fo test.fa.out
$ cat test.fa.out
>chr1:5-10
AAACC
2.2 将bam转为bed格式 =》 bamtobed
默认将bam转为6列的bed文件
$ bedtools bamtobed -i reads.bam | head -3
chr7 118970079 118970129 TUPAC_0001:3:1:0:1452#0/1 37 -
chr7 118965072 118965122 TUPAC_0001:3:1:0:1452#0/2 37 +
chr11 46769934 46769984 TUPAC_0001:3:1:0:1472#0/1 37 -
2.3 从bam中提取fastq =》 bamtofastq
$ bedtools bamtofastq -i NA18152.bam -fq NA18152.fq
2.4 12列bed转6列的bed =》 bed12tobed6
BED6表示文件包含bed格式中的前六列,是最为常见的格式; BED12表示文件包含bed格式中的所有12列,含有信息最为全面。
$ head data/knownGene.hg18.chr21.bed | tail -n 3
chr21 10079666 10120808 uc002yiv.1 0 - 10081686 1 0 1 2 0 6 0 8 0 4 528,91,101,215, 0,1930,39750,40927,
chr21 10080031 10081687 uc002yiw.1 0 - 10080031 1 0 0 8 0 0 3 1 0 2 200,91, 0,1565,
chr21 10081660 10120796 uc002yix.2 0 - 10081660 1 0 0 8 1 6 6 0 0 3 27,101,223,0,37756,38913,
$ head data/knownGene.hg18.chr21.bed | tail -n 3 | bed12ToBed6 -i stdin
chr21 10079666 10080194 uc002yiv.1 0 -
chr21 10081596 10081687 uc002yiv.1 0 -
chr21 10119416 10119517 uc002yiv.1 0 -
chr21 10120593 10120808 uc002yiv.1 0 -
chr21 10080031 10080231 uc002yiw.1 0 -
chr21 10081596 10081687 uc002yiw.1 0 -
chr21 10081660 10081687 uc002yix.2 0 -
chr21 10119416 10119517 uc002yix.2 0 -
chr21 10120573 10120796 uc002yix.2 0 -
tip:如果一个基因原来的BED12文件中一行记录有6个外显子,那么会将原来一行转为6行外显子信息存储在BED6中
2.5 合并坐标文件中的区间 =》merge
需要先排序:
sort -k1,1 -k2,2n in.bed > in.sorted.bed

# 说明:bedtools merge [OPTIONS] -i <BED/GFF/VCF/BAM>
$ cat A.bed
chr1 100 200
chr1 180 250
chr1 250 500
chr1 501 1000
$ bedtools merge -i A.bed
chr1 100 500
chr1 501 1000
# -d参数设置距离多远的两个坐标可以合并,比如上图的-d 10,就把input中相距10bp的两块合并了