210-RNA-Seq数据上游分析这么多pipeline,应该怎么选呢?
刘小泽写于2020.10.23
背景
随着芯片和NGS的应用越来越多, US Food and Drug Administration (FDA)启动了一个叫做:**MicroArray/Sequencing Quality Control (MAQC/SEQC)**的项目,旨在为数据分析提供一个标准和质量控制。
关于第一期 MAQC-I 【2006结束】
项目最早开始于2005年,即MAQC-I期,主要为芯片数据提供支持:
- provide quality control (QC) tools to the microarray community to avoid procedural failures
- develop guidelines for microarray data analysis by providing the public with large reference datasets along with readily accessible reference RNA samples
- establish QC metrics and thresholds for objectively assessing the performance achievable by various microarray platforms
- evaluate the advantages and disadvantages of various data analysis methods
后来到2014年又先后完成了II 、 III期,总共发表了约30篇文章,其中三分之一发在了Nature Biotechnology,而且其中两篇在过去十多年中保持了 Nature Biotechnology中的最高引用量
关于第二期 MAQC-II 【2010结束】
主要有两个目的:
- assess the capabilities and limitations of various data analysis methods in developing and validating microarray-based predictive models
- reach consensus on the “best practices” for development and validation of predictive models based on microarray gene expression and genotyping data for personalized medicine.
关于第三期 MAQC-III (also known as SEQC) 【2014结束】
开始偏向高通量测序方向的评测,而且更倾向于具体的技术应用可靠性
来自12个国家的73个组织中的180名研究人员,旨在:
- examine the latest tools for measuring gene activity (RNA-Seq)
- establish best practices for reproducibility across different technologies and laboratories
- evaluate the utility of these technologies in clinical and safety assessments.
测试了三个主流平台:Illumina HiSeq, Life Technologies SOLiD, and Roche 454
还测试了芯片与RNA-Seq工具的通用性
看看最新的第四期 MAQC-IV (also known as SEQC2)
项目主要有三个目的:
- (1) to develop quality metrics for reproducible NGS results from both whole genome sequencing (WGS) and targeted gene sequencing (TGS)
- (2) to benchmark bioinformatics methods for WGS and TGS towards the development of standard data analysis protocols
- (3) to assess the joint effects of key parameters affecting NGS results and interpretation for clinical application.
看一篇文章,对上游RNA-Seq的pipeline进行了排序
- 题目:Impact of RNA-seq data analysis algorithms on gene expression estimation and downstream prediction (Scientific Reports)
- 日期:2020.10.21
- 团队:Georgia Institute of Technology and Emory University
- 链接:https://www.nature.com/articles/s41598-020-74567-y
文章目的
Focused on the impact of the joint effects of RNA-seq pipelines on gene expression estimation as well as the downstream prediction of disease outcomes
使用的数据
- The FDA SEQC-benchmark dataset (Gene Expression Omnibus accession number GSE47792) :选取Beijing Genomics Institute (BGI) and Mayo Clinic (MAY)产出的数据,使用了4个样本(每个有4个重复)。同样SEQC提供了qPCR验证数据集(ncludes 20,801 genes assayed with PrimePCR. Each PrimePCR gene was assayed once for each of the four samples )
- SEQC-neuroblastoma dataset :176-sample neuroblastoma dataset
方法
- 提出评价指标: developed and applied three metrics (i.e., accuracy, precision, and reliability) to quantitatively evaluate each pipeline’s performance on gene expression estimation.
- 真实数据测试三个指标:investigated the correlation between the proposed metrics and the downstream prediction performance using two real-world cancer datasets
- 提供一些建议:provided scenarios as guidelines for users to use these three metrics to select sensible RNA-seq pipelines for the improved accuracy, precision, and reliability of gene expression estimation
- 这篇文章只提供了上游比对+定量+标准化的测试,下游R包的测试没有给出
发现
总共调查了278个RNA-Seq的pipeline( 附件说得很详细 Supplementary Information 1.)
上游的比对软件测试了bowtie、bowtie2、BWA、GSNAP、MapSplice、Novoalign、OSA、RUM、RUM、STAR、Subread、TopHat、WHAM,但没有常用的hisat2
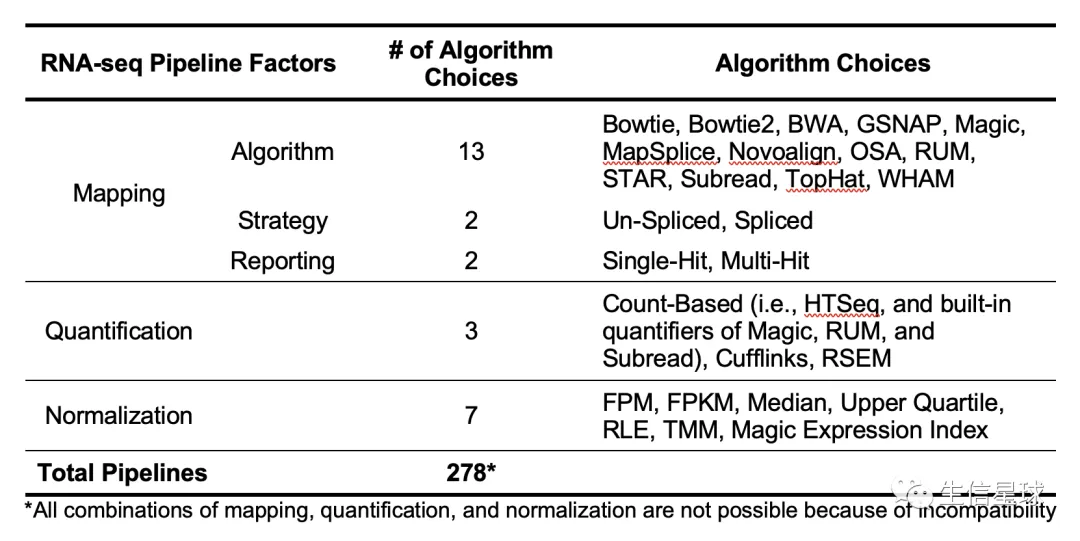
标准化的方法有:

测试结果中:
红色的Median normalization 在众多出色的pipeline中出现次数很多
Bowtie2 multi-hit and GSNAP un-splice 准确性最好

针对不用研究人群给出了三种使用场景的建议:

最后附件还给出了每个软件的用法:
##############################
# Mapping
##############################
# 1. Bowtie (with RSEM quantification)
#Index Preparation
rsem-prepare-reference --gtf [GTF File] [Genome Fasta] [Output Prefix]
#Mapping and Quantification
rsem-calculate-expression --paired-end -p [CPUs] --bowtie-path [Path to Bowtie] --phred33/64-quals --no-bam-output Read_1.fastq Read_2.fastq [RSEM Index] [Output Prefix]
# 2. Bowtie2
#Index Preparation
bowtie2-build [Reference Fasta File] [Reference Index Output]
#Single-Hit Mapping
bowtie2 -p [CPUs] -x [Reference Index (either AceView or HG)] -1 [read1.fastq] -2 [read2.fastq] > [output file.sam]
#Multi-Hit Mapping
bowtie2 -p [CPUs] -k 200 -x [Reference Index (either AceView or HG)] -1 [read1.fastq] -2 [read2.fastq] > [output file.sam]
# 3. BWA
#Index Preparation
bwa_index [Reference Fasta File]
#Mapping
bwa aln -1 -t [CPUs] [Reference Index (either AceView or HG)] read1.fastq > output1.sai
bwa aln -2 -t [CPUs] [Reference Index (either AceView or HG)] read2.fastq > output2.sai
bwa sampe [Reference Index (either AceView or HG)] output1.sai output2.sai read1.fastq read2.fastq > output.sam
# 4.GSNAP
#Index Preparation
gmap_build -D [Reference Index Output Directory] -d [Reference Index Output Name] [Reference Fasta File]
#Spliced Index Preparation (for Spliced Mapping)
iit_store -o transcriptome.iit [Reference Transcriptome GTF]
#Single-Hit Un-Spliced Mapping
gsnap --nthreads [CPUs] -B 5 -D [Reference Index Directory] --format=sam -n 1 -J 33 -d [Reference Index Name] read1.fastq read2.fastq > output.sam
#Multi-Hit Un-Spliced Mapping
gsnap --nthreads [CPUs] -B 5 -D [Reference Index Directory] --format=sam -n 200 -J 33 -d [Reference Index Name] read1.fastq read2.fastq > output.sam
#Single-Hit Spliced Mapping
gsnap --nthreads [CPUs] -B 5 -D [Reference Index Directory] --format=sam -n 1 -J 33 --d [HG Reference Index Name] -s [Transcriptome Spliced Index (*.iit)] --nofails read1.fastq read2.fastq > output.sam
#Multi-Hit Spliced Mapping
gsnap --nthreads [CPUs] -B 5 -D [Reference Index Directory] --format=sam -n 200 -J 33 --d [HG Reference Index Name] -s [Transcriptome Spliced Index (*.iit)] --nofails read1.fastq read2.fastq > output.sam
# 5.MapSplice
#Index Preparation
Same as Bowtie
#Mapping (SEQC-benchmark Data)
python mapsplice_segments.py -u Read_1.fastq,Read_2.fastq -B [Bowtie Genome Index] -c [Per Contig Genome FASTA Files] -o [Output Directory] [Configuration File]
#Configuration File:
reads_format = FASTQ
segment_mismatches = 1
segment_length = 25
read_length = 100
paired_end = yes
junction_type = non-canonical
full_running = yes
anchor_length = 8
remove_temp_files = yes
remap_mismatches = 2
splice_mismatches = 0
min_intron_length = 70
max_intron_length = 500000
threads = [CPUs]
search_whole_chromosome = no
map_segment_directly = no
run_MapPER = no
do_fusion = no
do_cluster = no
#Mapping (SEQC-application, Neuroblastoma data)
python mapsplice -p [CPUs] -o [Output Directory] --min-intron 70 --max-intron 500000 --bam -c [Per Contig Genome FASTA Files] -x [Bowtie Genome Index] -1 Read_1.fastq -2 Read_2.fastq
# 6.Novoalign
#Index Preparation
novoindex -t [CPUs] [Reference Output Index (*.ndx)] [Reference Fasta File]
#Single-Hit Mapping
novoalign -c [CPUs] -F ILMFQ -o SAM -r Random -d [Reference Index] -f read1.fastq read2.fastq > output.sam
#Multi-Hit Mapping
novoalign -c [CPUs] -F ILMFQ -o SAM -r all -e 200 -d [Reference Index] -f read1.fastq read2.fastq > output.sam
# 7.OSA
#Index Preparation
osa.exe --buildref [Base_Dir] [genome_fasta_file_name] [ref_lib_prefix]
osa.exe --buildgm [Base_Dir] [gtf_file_name] [ref_lib_prefix] [gene_model_prefix]
#Mapping
mono osa.exe --alignrna [OSA Index Directory] [Reference Genome Prefix] [Genome Annotation Prefix] [Configuration File]
#Configuration File:
<Files>
Read_1.fastq.gz
Read_2.fastq.gz
<Options>
ThreadNumber=CPUs
PairedEnd=True
FileFormat=FASTQ
AutoPenalty=True
FixedPenalty=2
Gzip=True
ExpressionMeasurement=TPM
SearchNovelExonJunction=False
GenerateSamFiles=False
WriteReadsInSeparateFiles=False
InsertSizeStandardDeviation=70
ExpectedInsertSize=170
ExcludeUnmappedInBam=True
<Output>
OutputName=OSA_ILM
OutputPath=XYZ
# 8.RUM
#Index Preparation
perl create_indexes_from_ucsc.pl NAME_genome.txt NAME_refseq_ucsc
#Mapping
rum_runner align -i [RUM Genome Index] -o [Output Directory] --name [Sample Name] --chunks [CPUs] Read_1.fastq Read_2.fastq
# 9.STAR
#Index Preparation
STAR --runThreadN 24 --runMode genomeGenerate --genomeDir aceview_seqc --genomeFastaFiles [HG Reference Fasta] --sjdbGTFfile [Transcriptome Reference GTF] --sjdbOverhang 100
#Mapping
STAR --genomeDir [STAR Genome Index] --readFilesIn [Read_1.fastq.gz] [Read_2.fastq.gz] --runThreadN [CPUs] --readFilesCommand zcat --outFileNamePrefix [Output Prefix] --outFilterMultimapNmax 200 --outSAMstrandField intronMotif --outFilterIntronMotifs RemoveNoncanonical --genomeLoad LoadAndKeep
# 10.Subread
#Index Preparation
subread-buildindex -B [HG Reference Index Name] [HG Reference Fasta]
#Mapping
subread-align -r read1.fastq -R read2.fastq -T [CPUs] -P 6 -u -H -i [HG Reference Index] -o output.sam
#featureCounts Quantification
featureCounts -p -T [CPUs] -t exon -g gene_id -a [Transcriptome GTF] -o output.counts input.sam
# 11.TopHat
#Index Preparation
Same as Bowtie2
#Single-Hit Mapping
tophat -p [CPUs] -o [Output Directory] --max-multihits 1 --library-type fr-unstranded --solexa1.3-quals --no-novel-juncs -G [GTF File] --transcriptome-index [Transcriptome Reference Index] [HG Reference Index] [Read_1.fastq.gz] [Read_2.fastq.gz]
#Multi-Hit Mapping
tophat -p [CPUs] -o [Output Directory] --max-multihits 200 --library-type fr-unstranded --solexa1.3-quals --no-novel-juncs -G [GTF File] --transcriptome-index [Transcriptome Reference Index] [HG Reference Index] [Read_1.fastq.gz] [Read_2.fastq.gz]
# 12.WHAM
#Index Preparation
wham-build -l 90 [Reference Fasta File] [Reference Index Output]
#Single-Hit Mapping
wham -l 90 -g 1 -X 500 --best -S -t [CPUs] -k 1 -1 read1.fastq -2 read2.fastq [Reference Index] output.sam
#Multi-Hit Mapping
wham -l 90 -g 1 -X 500 --best -S -t [CPUs] -k 200 -1 read1.fastq -2 read2.fastq [Reference Index] output.sam
##############################
# Expression Quantification
##############################
# 1. HTSeq
samtools view -h [BAM File] | htseq-count --mode=intersection-nonempty --stranded=no --type=exon --idattr=gene_id - [GTF File] > [Output File]
# 2. Cufflinks
cuffdiff -o [Output Directory] -p [CPUs] -b [Reference Genome FASTA] -u -L [Sample Labels] [GTF Files] [BAM Files]
# 3. RSEM
rsem-calculate-expression --bam --paired-end -p [CPUs] --no-bam-output [BAM File] [RSEM Index] [Output Prefix]