026-转录组差异基因筛选
刘小泽写于18.8.24~简单说说这几天的体会吧
Linux的部分结束了,主要就是比对、定量两大任务; 然后开始使用R语言进行下游分析,首先筛选差异基因,然后绘制各种图,于是最近开始学习R,之前有接触,因为我是喜欢可视化的,喜欢美的事物,因此学了ggplot2还有一些简单的R操作,但由于长时间没有需求,就没有深入学习。现在通过一些分析发现,后面确实离不开R,没有linux可以用R分析,但是没有R,真的不好弄啊。重新学习发现,R有大部分是现成的,只要你能找到源代码,自己稍加修改就能用,不用绞尽脑汁去自己想怎么实现
但说到底,代码、图终究不是目的,还是要解决实际生物问题才是真理;另外只有自己掌握技术,才能有创新的可能
#学习了bioconductor的DEG包(https://www.bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html#standard-workflow)
#以及jimmy的R教程(https://github.com/jmzeng1314/my-R/blob/master/10-RNA-seq-3-groups/hisat2_mm10_htseq.R)都特别给力
rm(list = ls()) #首先清空R中的变量
options(stringsAsFactors = F) #在读入数据时,遇到字符串之后,不将其转换为factors,仍然保留为字符串格式
标准的表达矩阵求相关性
以airway数据包为例
library(airway)
data(airway) #airway就是一个数据包,只是为了提供数据
#如何取它的表达矩阵呢?一般使用exprs()来取
exprs(airway) #但这里这样做是报错的
#可以先看一下airway 是什么东西
airway #结果显示,他的class是RangedSummarizedExperiment
#这样,可以使用?RangedSummarizedExperiment 来查看怎样取表达矩阵
#看帮助信息中的示例文件,发现head(assay(rse)),所以他可能是利用assay取的表达矩阵
exprMtx=assay(airway)
png('cor_3.png')
corrplot(cor(log2(exprMtx_3+1)))
dev.off() #结果发现相关性都是接近1
png('heatmap3.png')
m3=cor(log2(exprMtx_3+1))
pheatmap(scale(m3))
dev.off()
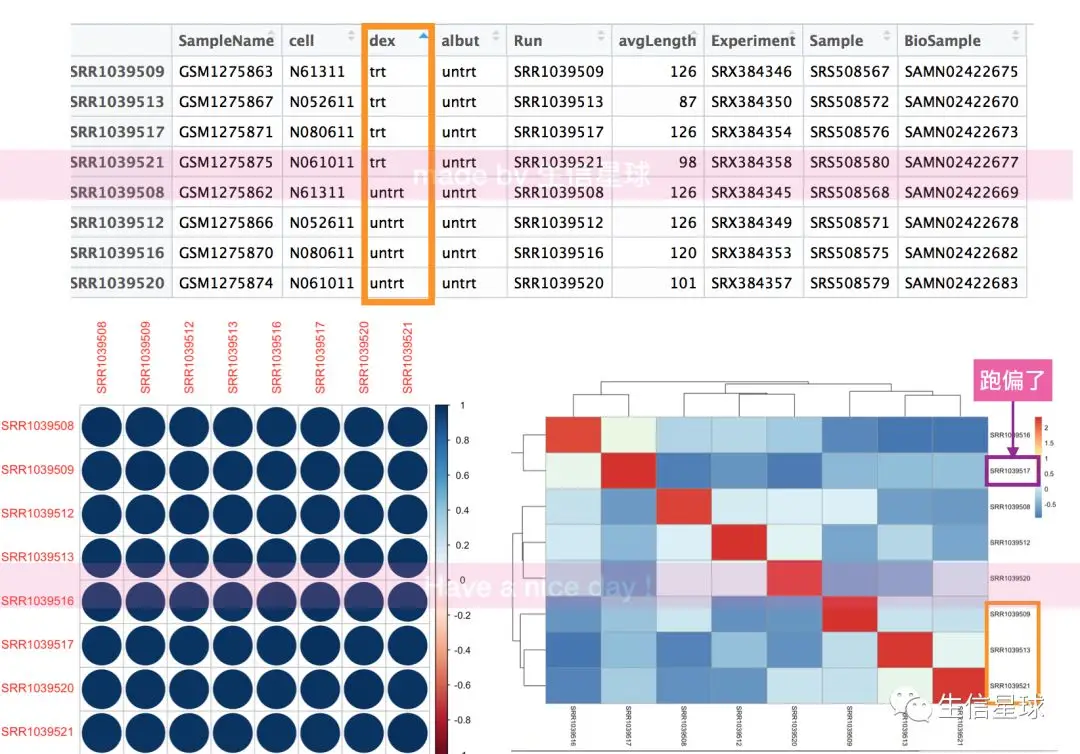
#如何判断这个图准不准呢?看原始表达矩阵,使用colData()
tmp=colData(airway)
#然后按照第三列dex排序,4个trt,4个untrt(08,12,16,20),发现有一个跑偏了
标准化基因名(可选)
示例joint.count公众号后台回复joint.count即可获取,用来测试
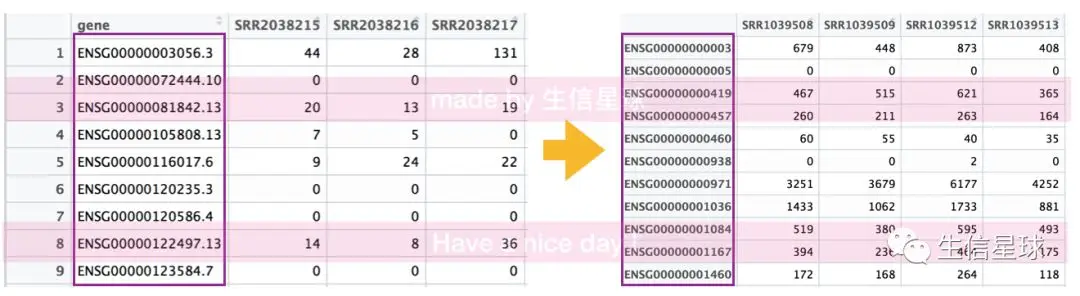
#定量结果得到的基因名可能是这样的ENSG00000072444.10,但是在Ensembl数据库中搜索,只能搜索标准命名ENSG00000072444,后面的.10搜不到,因此需要去除
c=read.table('joint.count',header = T)
#开始转换第一列基因名(去小数点)
install.packages("stringr")
library(stringr) #利用这个stringr包,对字符串进行处理
#按gene名的小数点分隔,我们要小数点前的部分,也就是分隔后的第一列
c$gene = str_split(c$gene, '\\.',simplify = T)[,1]
#假如是有好几个单独的文件,想合并在一起,就用merge
# merge(x1,x2,by='gene')
DESeq2进行差异分析
官网:https://www.bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html#standard-workflow
#没有安装过DESeq2的先安装这个
source("https://bioconductor.org/biocLite.R")
biocLite("DESeq2")
suppressMessages(library(DESeq2)) #suppressMessages加载包时不显示提示信息
library(airway) #这个包里包含了提取表达量的assay、获得列信息的colData等函数
data(airway)
#group=colData(airway) #得到airway全部组名
group_list=colData(airway)[,3] #得到样本分组(也就是第三列,trt、untrt)
exprMtx=assay(airway) #assay提取表达量
#一个数据框需要行和列,将行名命名为表达量的列名,也就是SRR等样本名;列名是分组信息
colData=data.frame(row.names = colnames(exprMtx), group_list=group_list)
# 开始DESeq分析
# 查看官方操作代码1:dds <- DESeqDataSetFromMatrix(countData, DataFrame(condition), ~ condition)
# 代码2: DESeqDataSetFromMatrix(countData, colData, design)
#countData,就是我们上面构建好的表达量矩阵exprMtx;
#DataFrame(condition)这个东西首先一看是个数据框,而且是关于condition的数据框。我们这里的变量有样本号SRR、处理对照分组、表达量三种信息,表达量已经用了,那么数据框就应该用样本名和处理对照分组信息来构建,而且人家提示condition,也就是处理对照分组trt和untrt,就可以构建行名为样本号,列名为分组信息的数据框;上面建立的colData就是这个DataFrame(condition);
#~condition就是处理对照分组信息,上面构建的group_list
dds = DESeqDataSetFromMatrix(countData = exprMtx, colData = colData, design=~ group_list)
# 然后就能运行了
dds = DESeq(dds)
# 从deseq结果中提取出统计信息,比如平均值、log2FC、标准误、t检验、p-value等
# 官方给出的模式是:res <- results(dds, contrast=c("condition","treated","untreated")),我们只需要将condition、treated、untreated替换成我们这里存在的即可
res = results(dds, contrast=c("group_list","trt","untrt"))
# 但是res只是一个数据框对象,就是表达了它包含数据框的内容,如果直接看会比较复杂,因此需要再得到里面包含的矩阵
# 要得到差异基因,先调整p值,将res中p值从小到大排序
# 官网操作 resOrdered <- res[order(res$pvalue),]
resOrdered = res[order(res$pvalue),]
#看一下有多少p值是小于0.1的
sum(res$padj < 0.1, na.rm=TRUE)
#看一下前6行
head(resOrdered)
# 之前提到,不管res还是resOrdered都是对象的形式,如果要进行进一步分析,需要弄成一个数据框,使用as.data.frame将筛选的部分保存为数据框,命名为deg
deg=as.data.frame(resOrdered) #可以看到,得到的deg差异基因现在有64102个
#可以看到,得到的deg差异基因现在有64102个,但是有一个问题,这些deg中有可能会出现NA,毕竟我们是直接从原始统计数据中只排了个序,得到的结果有一部分是缺失值(NA,not available),而这部分是不能用来后续作图的
# 排除NA值
deg = na.omit(deg) #可以看到,排除后还剩18595个
画图
## 1.画一个热图
library(pheatmap)
#首先从过滤后的deg中选出p-value从小到大前50的基因名,保存到filt_gene中
filt_gene=head(rownames(deg),50)
#从表达矩阵中将这些基因的表达量筛出来,放到过滤的矩阵filr_mtx中
filt_mtx=exprSet[filt_gene,]
# 再使用scale()进行表达量标准化【注意:原来的filt_mtx中行为基因名,列为样本名】我们需要让列成为基因名,毕竟我们是想看不同基因间的差异;如果要看样本之间的差异,就不用转换了
# 假如像下面两行,直接标准化,做出的图就是针对样本间的,没什么差异性【这样就对了!样本间差异性大了,就说明实验有问题了;可以这么理解:样本要求一致性,基因要求差异性】
# 1
filt_mtx=scale(filt_mtx)
pheatmap(filt_mtx)
rm(filt_mtx)
#再次画图之前,要将之前的filt_mtx清空,再重新跑一遍
# 正确的做法:先对filt_mtx转置=》标准化=》再转过来【再转一次的目的就是改改横纵坐标,保持和之前一致,否则本来50个应该横向出现的基因,就会纵向出现】
# 2
filt_mtx=exprMtx[filt_gene,]
filt_mtx=scale(t(filt_mtx)) # 先来一个转换一次的
pheatmap(filt_mtx)
rm(filt_mtx)
#3
filt_mtx=exprMtx[filt_gene,]
filt_mtx=t(scale(t(filt_mtx))) # 再来一个转换两次的
pheatmap(filt_mtx,filename = 'DEG_pre50_heat.png')
