223-这么好的xena官方教程,不看可惜了
刘小泽写于2021.1.4 想必大家都听过或者用过xena这个网站,我主要是利用它下载数据,链接在:https://xenabrowser.net/datapages/ 其实它的网页工具做的也是非常的人性化,功能很多,而且不用自己下载数据,方便了不少同学。另外UCSC Xena 的国内镜像: https://xena.hiplot.com.cn/
首先看看它的大体功能
xena收集了来自各个癌症中心的数据,包括The Cancer Genome Atlas (TCGA), International Cancer Genome Consortium (ICGC), Genomic Data Commons (GDC), and UCSC RNA-seq compendium(http://xena.ucsc.edu/public)

也正是因为这么大量数据的加持,xena可以完成 SNVs, INDELs, large structural variants, copy number variation, gene-, transcript-, exon-, miRNA-, LncRNA-, protein-expressions, DNA methylation, ATAC-seq signals, phenotypic annotations等等分析
分析的界面长下面这样:

右下角也标注了:
- 如果分析涉及了tumor与normal的比较,那么可以用
TCGA TARGET GTEx - 如果要查看非编码突变,可以用
ICGC的数据
另外,有一个有趣的功能就是:可以把自己分析完的数据保存为网页链接,分享给别人,打开可以跳转到你分析的结果
除此以外,还有大量的详细教程
丰富的视频、PPT、文本资源尽在此:
https://ucsc-xena.gitbook.io/project/tutorials
下面跟着其中一个PPT教程走一遍
https://ucsc-xena.gitbook.io/project/tutorials#advanced-slide-tutorial
1 可以用Xena帮助解决什么问题
- 某个基因的高表达与预后的关系?
- 自己定义的两个分组之间的生存存在差异吗?
- 某个基因在三种癌症类型的tumor与normal样本是不是存在差异表达?
- 某个基因的突变、拷贝数变异、基因表达量之间存在怎样的关系?
2 Xena中存储的公共数据
TCGA
- 40种癌症类型,11000样本
- 主要是primary tumor,还有少量的normal和metastatic
- 数据类型主要有:somatic mutation、gene expression、copy number
- 生存及其他基础表型、临床数据(如年龄、亚型)
TARGET
https://ocg.cancer.gov/programs/target
全称是:The Therapeutically Applicable Research to Generate Effective Treatments (TARGET)
主要围绕儿童开展,包括以下项目,和TCGA类似都有转录组、WGS、WES、甲基化等:
- Acute Lymphoblastic Leukemia
- Acute Myeloid Leukemia
- Kidney Tumors
- Neuroblastoma 神经母细胞瘤
- Osteosarcoma 骨肉瘤
- Pan-cancer Model Systems
TCGA TARGET GTEx
GTEx全称Genotype-Tissue Expression,存储了正常组织的表达量数据
ICGC
ICGC 全称International Cancer Genome Consortium(国际癌症基因组联盟),有亚洲、澳大利亚、欧洲、北美和南美的数据
PCAWG
全称是:Pan-Cancer Analysis of Whole Genomes(泛癌全基因组分析),是TCGA工作人员利用TCGA数据当中的WGS数据进行的分析,有2700多个样本供体
CCLE
https://portals.broadinstitute.org/ccle
Cancer Cell Line Encyclopedia(癌症细胞系百科全书),有各个人种肿瘤细胞系的WES数据,WGS数据,RNAseq数据,扩增子数据。可以找基因表达变化、突变、indels、拷贝数变异、甲基化、药物反应等等。目前已有1,457个 细胞系,涉及84,434个基因
MET500
https://met500.path.med.umich.edu/
主要是研究转移癌
未来还会加入蛋白组学的数据 ,来自:Clinical Proteomic Tumor Analysis Consortium (CPTAC)
3 常见的癌症可视化类型
体细胞突变 somatic mutation
DNA测序得到,主要包括:snv、indel这样的小型突变,对蛋白质的影响主要表现在:
- 沉默突变 Silent:不改变蛋白质
- 错义突变 Missense:一个氨基酸被另一个氨基酸取代
- 剪切位点突变 Splice-site:干扰剪切过程
- 有害突变 Deleterious:无义突变(nonsense)、移码突变(frameshift)
**无义突变:**单个碱基的替换引起出现了终止密码子,从而提前终止了多肽链的合成 **移码突变:**在一条DNA链上缺失或者插入1个、2个或者其他非3个及其整数倍的碱基,就会引起作用部位之后的密码子的组成及顺序发生变化,从而导致终止码提前或者延后
拷贝数变异 CNV
DNA测序得到,也是体细胞突变。主要分为扩增(amplification)和缺失(deletion)
基因表达数据
RNA测序得到,主要有转录本层面、基因层面、外显子层面、编码蛋白基因层面、非编码RNA层面
需要注意批次效应(由于实验方法、文库制备、分析方法等等产生),因此在比较不同数据集时要当心
需要注意
不是所有的样本都有全部的测序数据,比如某种癌症的某个样本某种测序质量差、文库制备出了问题,它就会缺失这种测序结果
4 支持的数据分析种类

!当心!虽然都是TCGA,但数据也存在差异
- 首先是:TCGA PanCan Atlas,它采用的是TCGA最高质量的数据,采用了统一的pipeline进行分析,大大减小了系统误差;附带了一些衍生数据,比如stemness score、immuno subtype;另外它的生存数据也是经过人工核验的;比对过程是比对到了2006 genome (hg19);在xena中的名称是:TCGA Pan-Cancer (PANCAN);适用于:全新的分析
- 其次是:GDC TCGA data,它也是统一分析,数据来自Genomic Data Commons(GDC);比对到了2013 genome(hg38);没有其他衍生数据,只是level3的基因组数据;在xena中的名称是:GDC xxx,如 GDC TCGA Bile Duct Cancer (CHOL);适用于:单个癌症(不想从pan cancer数据取子集)或者需要hg38版本的数据
- 最后是:最原始的TCGA data ,TCGA官方放出来的原始数据;大部分是比对到hg19,但也有比对到hg18的;在xena中的名称是:TCGA xxx,如TCGA Bile Duct Cancer(CHOL);适用于:需要外显子表达量的分析或者需要重复一些早期的工作
目前比较推荐的数据使用方式

5 可视化简单上手——以肺癌的EGFR为例
xena的可视化逻辑就像是excel表格一样,一列一列把癌症样本、想要分析的数据类型、基因等等加载进来
5.1 了解一下这个基因EGFR
- 10-35%的肺癌病人中会存在EGFR基因的异常(突变或扩增)
- 在女性中最常见
- EGFR抑制剂目前在临床上已经应用,比如EGFR酪氨酸激酶抑制剂(EGFRTKI)(例如埃罗替尼和吉非替尼)就是一类EGFR阻断药物,在治疗EGFR突变的肺癌中是有效的。目前前三代都出现了抗药问题,来自纪念斯隆-凯特琳癌症中心的研究显示,奥希替尼的初治和后线治疗展现了不同的耐药谱(https://med.sina.cn/article_detail_103_2_90349.html)
5.2 加载癌症数据
加载进来后,看到每一行表示一个样本
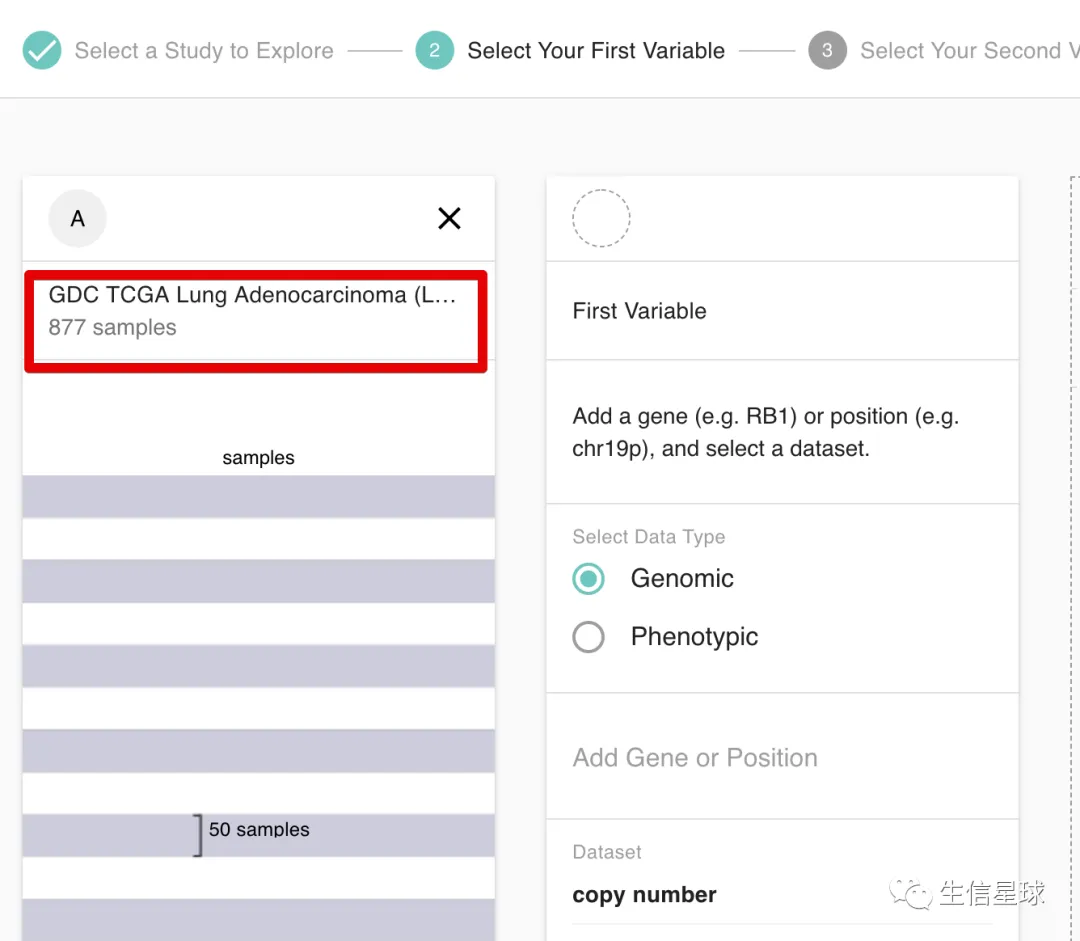
5.3 载入EGFR表达量
B列的每一行也是和A保持一致。null表示该样本没有数据,显示灰色

5.4 载入EGFR体细胞突变数据
C列中,突变位点就是其中的散点,不同颜色表示不同突变影响
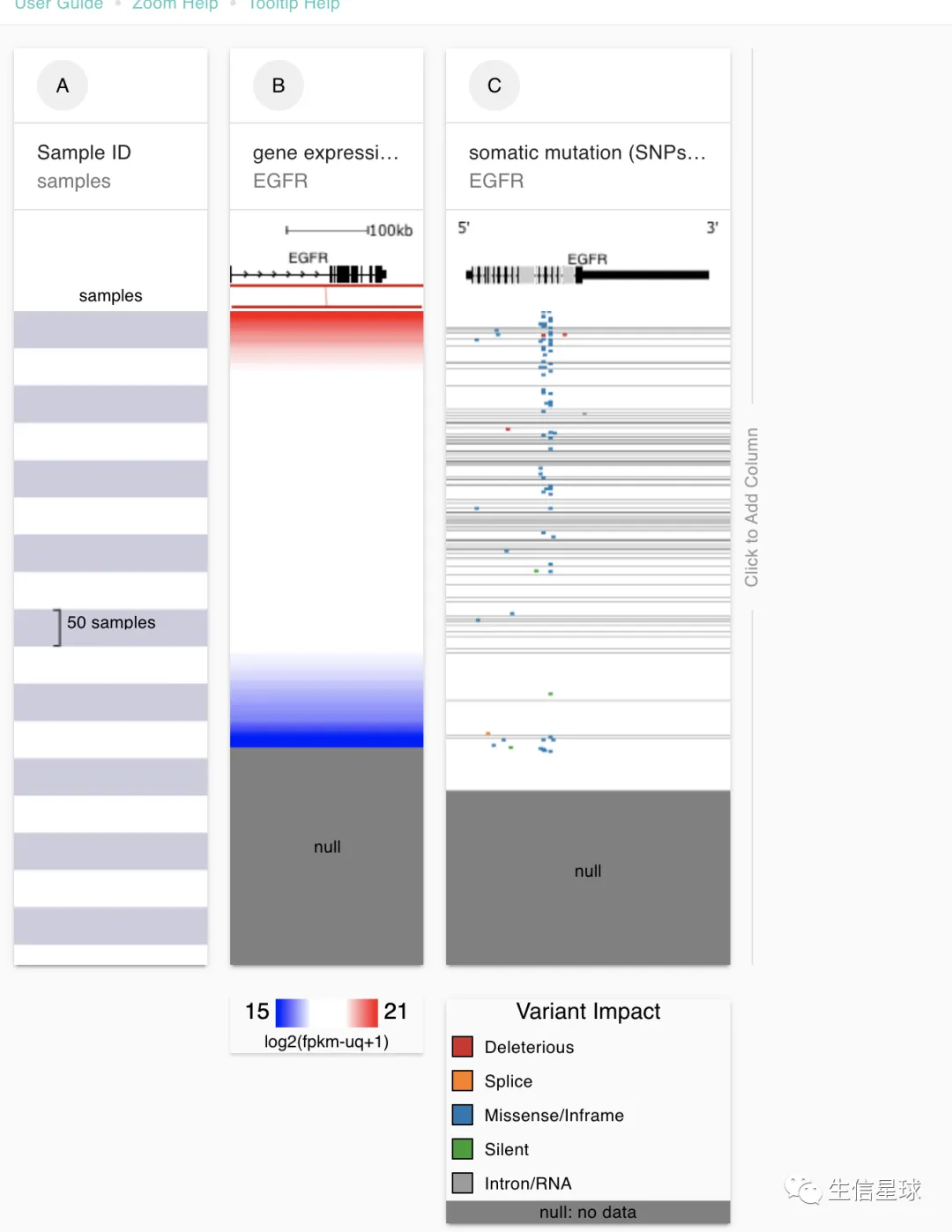
5.5 载入EGFR的拷贝数变异
默认只能加三列,此时需要点击Click to Add Column来增加。红色是扩增,蓝色是缺失

5.6 在上图发现,EGFR有异常的地方,表达量也比较高=》思考:它的异常对生存有没有影响?
思路是: step1- 去掉空值(Null)的样本 step2- 分组,一组有突变,一组没有 step3- 做生存分析
xena样本过滤小技巧:

step1: 过滤条件为!=null ,过滤完空值的样本,剩下508个

step2: 怎么定义EGFR异常呢?
根据上图看到,EGFR的异常(Aberration)可以包括:
- mutation:missense OR inframe deletion
- copy number:copy number gain
因此过滤条件就是:(mis OR infra) OR D:>0.5
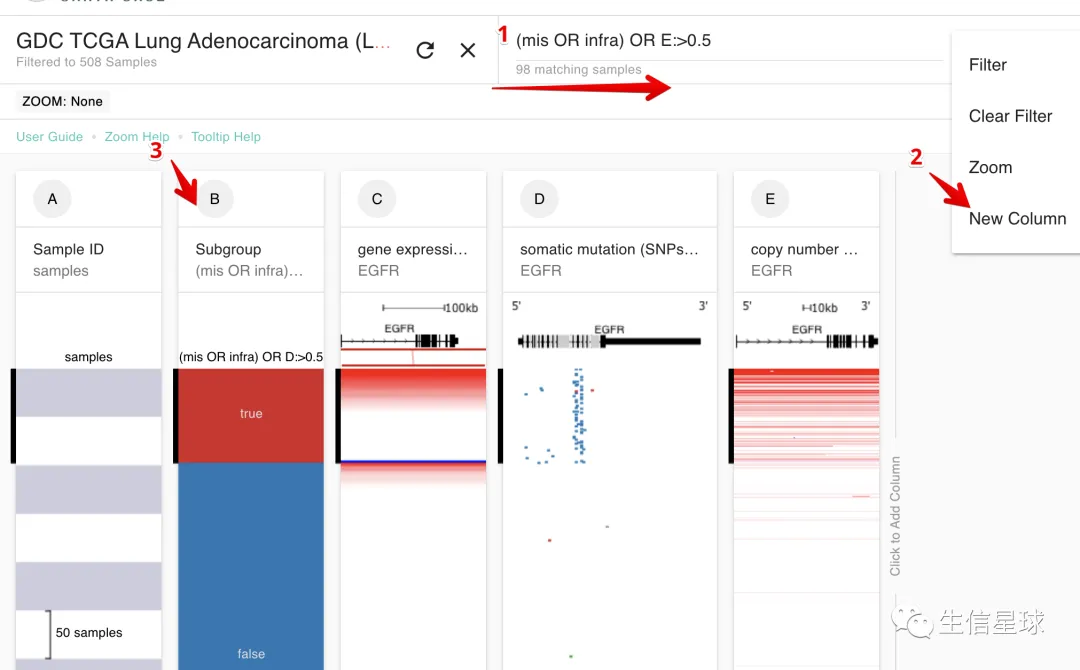
step3:怎么做生存分析呢?
5.7 表达量差异在自己分的两组之间差异显著吗?
选择KM plot下面的👇那个Chart & Statistics ,把X、Y轴设置一下就ok
