236-给你几个基因的氨基酸位点,如何拿到对应的基因组坐标?
刘小泽写于2021.3.1 最近接到这么一个需求,乍一看上去摸不着头脑,但仔细想想,却是一环扣一环
先看示例文件
这是一个我精简后的测试文件,一共三列,分别是:基因、蛋白ID、氨基酸位点
hg_Gene_name hg_Protein_accession hg_pos
BRD4 O60885 1064
NFRKB Q6P4R8 351
SAP30 O75446 131
SIK3 Q9Y2K2 551
SOX5 P35711 138

比如第一行:我想看BRD4这个基因的第1064个氨基酸,对应的基因组坐标是多少?
思考
如果想知道氨基酸在基因组上的坐标,那么首先我要知道这个基因的坐标,以及它的正负链信息【注意:正负链这里很重要,不要忽视】
一个氨基酸就是3个碱基,所以
要查的氨基酸对应的第一个碱基坐标 = 编码起始坐标 + 3* (氨基酸位点 - 1)。比如第2个氨基酸对应的第一个碱基坐标就等于:编码起始坐标 + 3 * (2 -1 )。因此,如何拿到一个基因的编码起始坐标就至关重要然后要明确基因结构,因为坐标的获取并不是从第一个碱基开始数,而从编码区开始
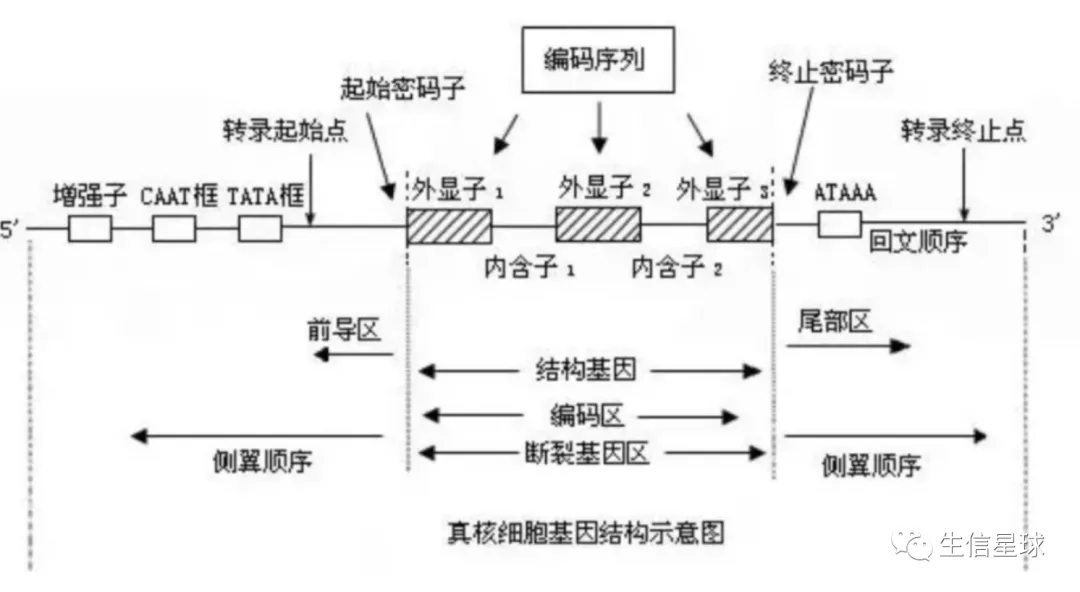
如何获取基因的编码区域?我这里采用的是借助NCBI 的 CCDS文件
首先获取hg38全部基因坐标和正负链信息
这里介绍两种方法,一个是UCSC的
TxDb注释包;一个是Ensembl的biomart函数。我更推荐biomart,因为转换信息实在是太丰富了!当然,hg19也是类似的
#--- 获取hg38 gene range ---#
# 方法一:根据UCSC
library(org.Hs.eg.db)
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg38.knownGene
genes_txdb=as.data.frame(genes(txdb))
s2g=toTable(org.Hs.egSYMBOL)
genes_hg38=merge(genes_txdb,s2g,by='gene_id')
head(genes_hg38)
colnames(genes_hg38)=paste0('hg38_',colnames(genes_hg38))
rownames(genes_hg38)=genes_hg38$hg38_symbol
hg38_ucsc=genes_hg38
# 方法二:根据ensembl
library("biomaRt")
ensembl <- useMart("ensembl", dataset = "hsapiens_gene_ensembl")
# attributes <- listAttributes(ensembl)
# View(attributes)
attr <- c("ensembl_gene_id","hgnc_symbol","chromosome_name",'start_position','end_position','strand')
ids <- getBM(attributes = attr,
filters = "hgnc_symbol",
values = genes_hg38$hg38_symbol,
mart = ensembl)
hg38_enb=ids
hg38_enb=hg38_enb[!grepl('CHR',hg38_enb$chromosome_name),]
hg38_enb=hg38_enb[!duplicated(hg38_enb$hgnc_symbol),]
colnames(hg38_enb)=paste0('hg38_',colnames(hg38_enb))
rownames(hg38_enb)=hg38_enb$hgnc_symbol

把这些信息先加进来
test=rio::import('test_AA.xlsx')
new2=cbind(hg38_enb[test$hg_Gene_name,3:6],test)
colnames(new2)[1]='chr'
colnames(new2)[2]='hg38_gene_start'
colnames(new2)[3]='hg38_gene_end'
colnames(new2)[4]='strand'
> head(new2)
chr hg38_gene_start hg38_gene_end strand hg_Gene_name hg_Protein_accession hg_pos
BRD4 19 15235519 15332545 -1 BRD4 O60885 1064
NFRKB 11 129863636 129895590 -1 NFRKB Q6P4R8 351
SAP30 4 173369969 173377532 1 SAP30 O75446 131
SIK3 11 116843402 117098437 -1 SIK3 Q9Y2K2 551
SOX5 12 23529504 24562544 -1 SOX5 P35711 138
怎么拿到基因的编码区域坐标呢?
以BRD4基因为例,我在https://www.ncbi.nlm.nih.gov/gene/23476下载了它的全部序列,密密麻麻,和IGV对比发现是一致的。
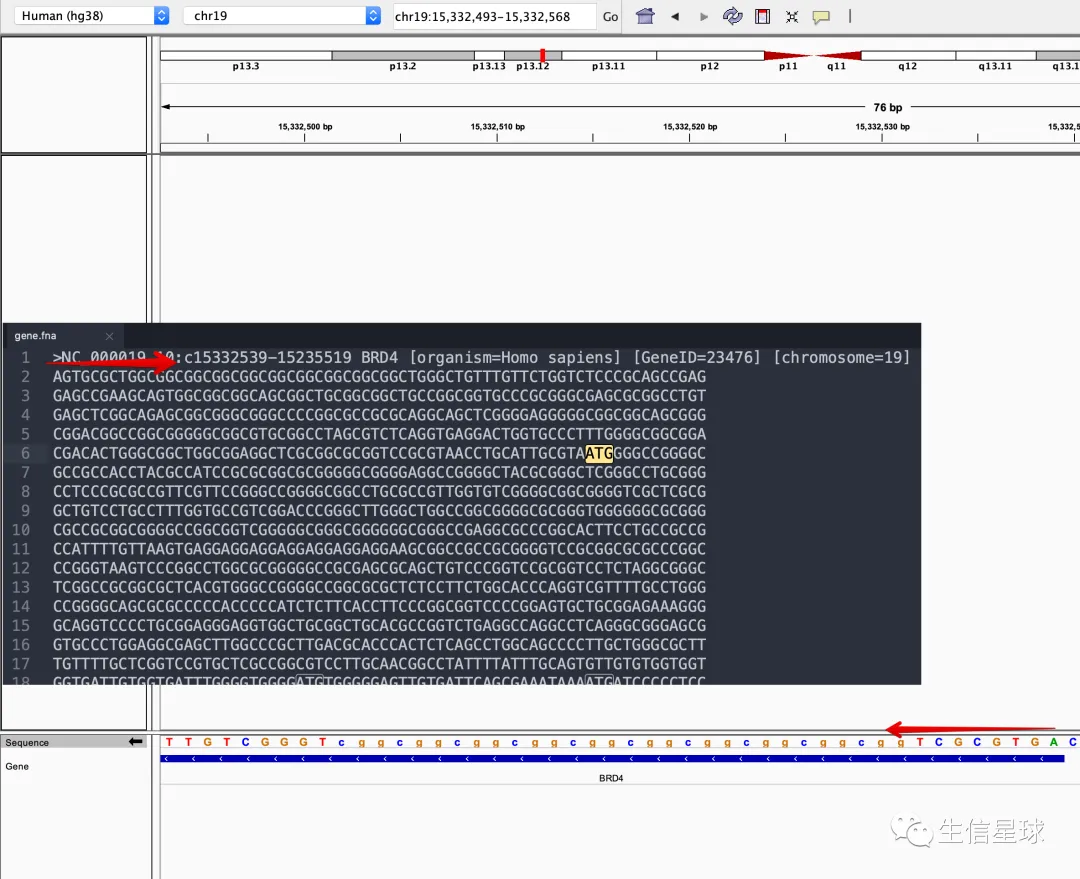
我们知道起始密码子对应的DNA序列是ATG,但是搜索ATG你会发现很多很多,那怎么到底从哪里开始算呢?其实这个不需要我们关心,只需要拿到CCDS文件
https://ftp.ncbi.nlm.nih.gov/pub/CCDS/current_human/CCDS.current.txt
然后看到其中就有基因以及它的CDS起始、终止,以及各个外显子的区域
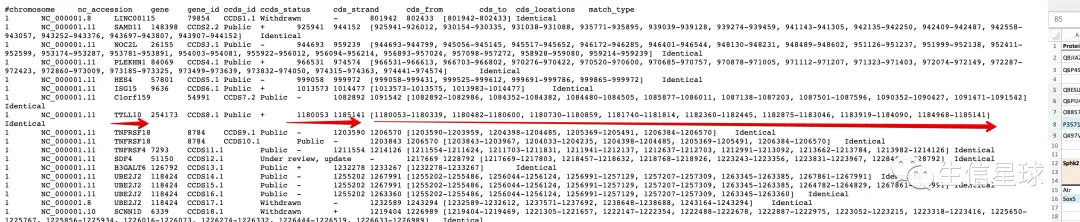
需要注意,CCDS数据库是基于hg38坐标的:
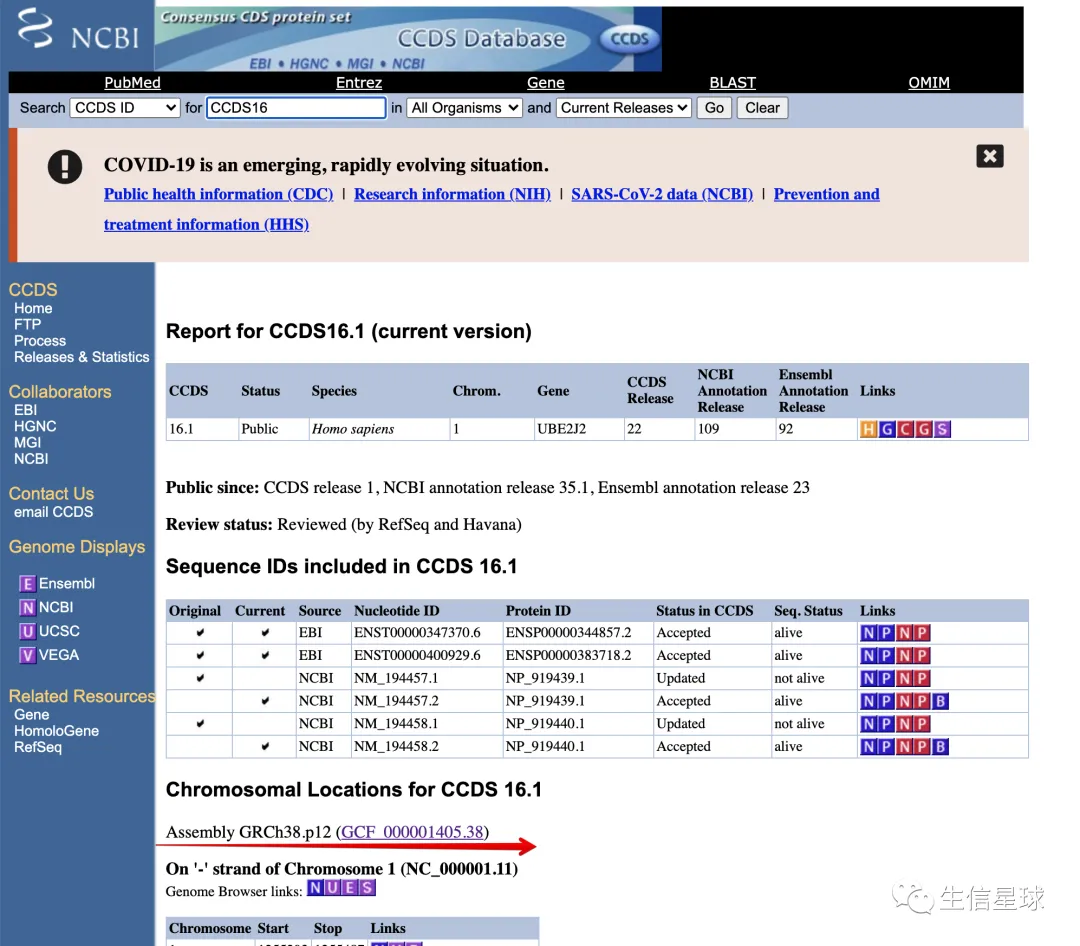
这时你可能会想,只要拿到
cds_from这一列,不就等于拿到了起始坐标?
但是,cds_to直接减去cds_from并不是实际的CDS区域,这个数值会大于真实值。我们应该用的,其实是最后一列
ccds=rio::import('CCDS.current.txt')
ccds=ccds[,c(1,3,8:10)]
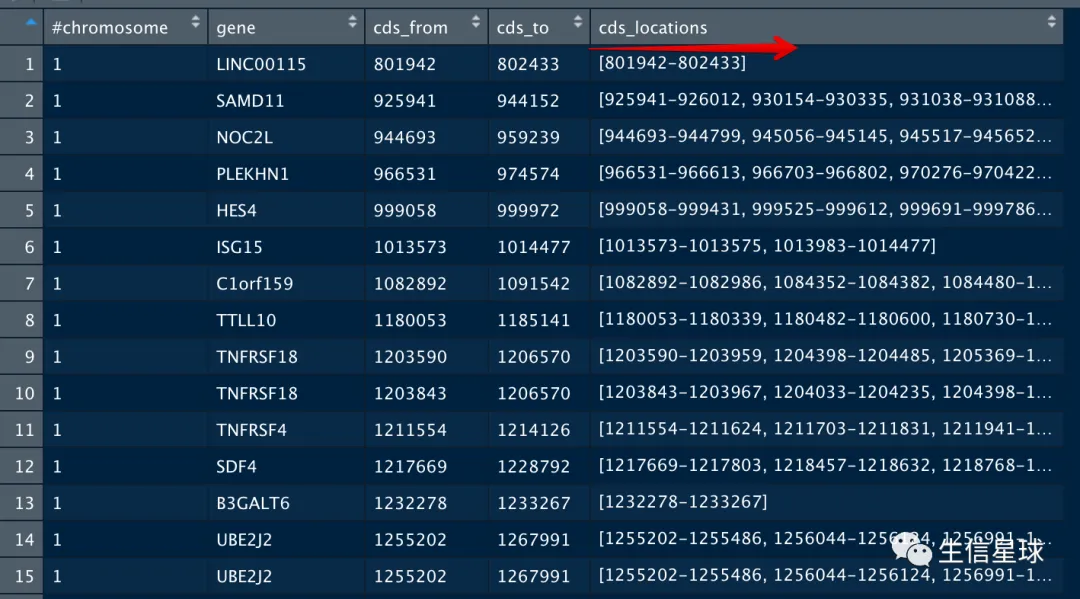
下面介绍2个tip
tip1 CCDS.current.txt文件中的坐标需要加1
如图:上图是CCDS文件给的结果,下图是CCDS数据库(https://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi?REQUEST=CCDS&DATA=CCDS12328)给的BRD4基因的结果
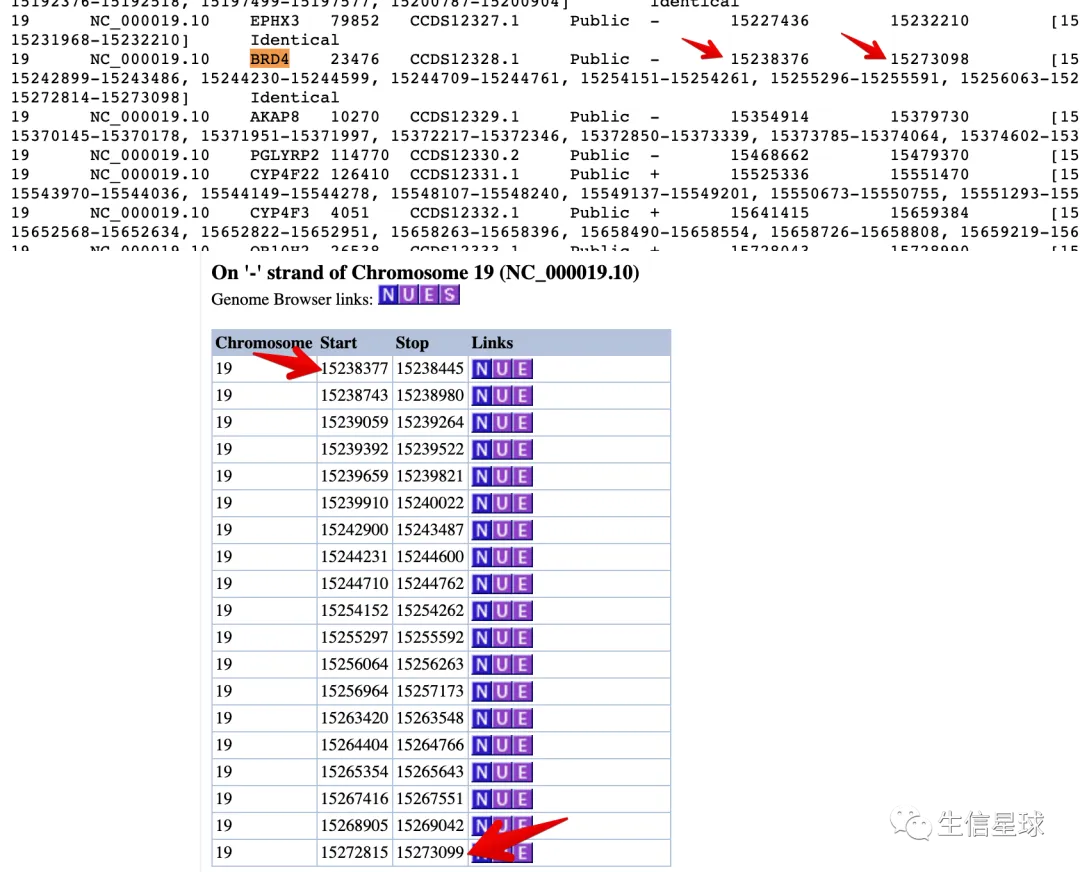
# 先把起始、终止坐标都加1
x=ccds[ccds$gene%in%new2$hg_Gene_name,]
x$cds_from=as.numeric(x$cds_from)+1
x$cds_to=as.numeric(x$cds_to)+1
tip 2 获取全部外显子的碱基坐标
试想:如果我拿到了一个基因全部外显子的碱基坐标,那么我想取第100-102个碱基,是不是很轻松就能拿到对应的坐标信息呢?那其实就是我们最后想要的氨基酸对应的基因组坐标。
要拿到一个基因全部外显子的碱基坐标,我写了下面的代码:
all_pos_list=lapply(x$cds_locations, function(x){
str_remove(x,'\\[') %>% str_remove(.,']') %>% str_split(.,',',simplify = T) %>%
apply(.,2,function(x){
# 记得将CCDS中的pos+1,才是真正的碱基位置
seq(str_split(x,'-',simplify = T)[1],str_split(x,'-',simplify = T)[2])+1
}) %>% unlist()
})
names(all_pos_list)=paste(x$gene,x$cds_from,x$cds_to,sep='-')
结果一目了然:

我之所以这么写,是因为在CCDS文件中,一个基因可能会对应多个转录本,因此一个基因会存在多行信息,我现在不想进行过滤,用name-start-end的方式把它们区分开来
最后,我把全部的信息merge到原来的new2中
ccds_dat=merge(x,new2,by.x = 'gene',by.y = 'hg_Gene_name',all.x = T)
ccds_dat=ccds_dat[,-5]
ccds_dat$id=paste(ccds_dat$gene,ccds_dat$cds_from,ccds_dat$cds_to,sep='-')
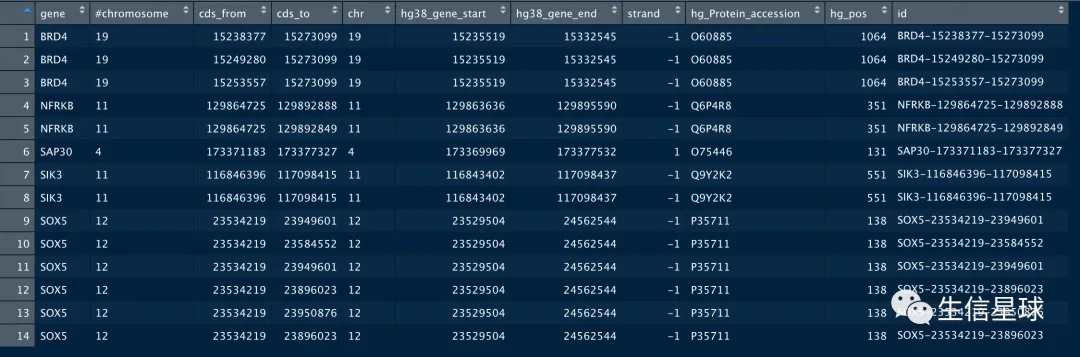
最后拿到坐标信息
这里需要考虑正负链
ccds_dat$check_aa_start=c()
ccds_dat$check_aa_end=c()
for(i in 1:nrow(ccds_dat)){
# i=4
name=ccds_dat$id[i]
if(ccds_dat$strand[i]==1){
ccds_dat[i,'check_aa_start']=all_pos_list[[name]][1+3*(ccds_dat[i,'hg_pos']-1)]
ccds_dat[i,'check_aa_end']=all_pos_list[[name]][1+3*(ccds_dat[i,'hg_pos']-1)]+2
}else if(length(all_pos_list[[name]])-3*(ccds_dat[i,'hg_pos']-1)>0){
ccds_dat[i,'check_aa_start']=all_pos_list[[name]][length(all_pos_list[[name]])-3*(ccds_dat[i,'hg_pos']-1)]-2
ccds_dat[i,'check_aa_end']=all_pos_list[[name]][length(all_pos_list[[name]])-3*(ccds_dat[i,'hg_pos']-1)]
}else{
# 有的转录本并不符合我们这里的pos信息,不管他
ccds_dat[i,'check_aa_start']=NA
ccds_dat[i,'check_aa_end']=NA
}
}
ccds_dat=ccds_dat[!is.na(ccds_dat$check_aa_start),]
ccds_dat=ccds_dat[,c(1:8,12:13,9:10)]

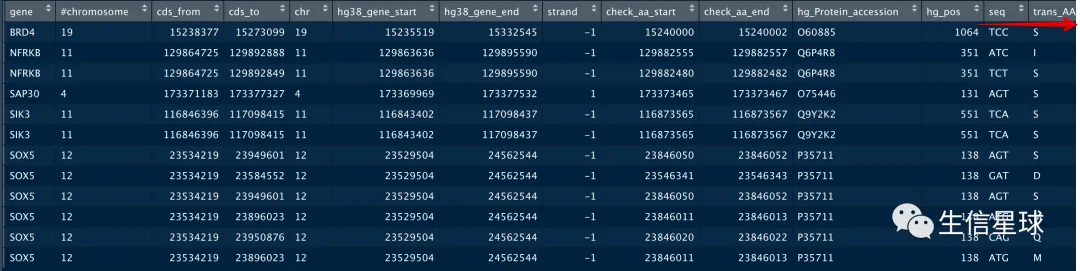
拿到坐标后,能做什么?
我们拿到了想要的氨基酸坐标,其实还可以把对应的序列取出,还能看看翻译后的氨基酸是不是符合我们预期,还可以根据序列查看有没有突变位点
这里举个例子:看看碱基以及翻译后的氨基酸信息
suppressMessages(library(GenomicFeatures))
suppressMessages(library(BSgenome.Hsapiens.UCSC.hg38))
library(Biostrings)
gr=GRanges(seqnames = paste0('chr',ccds_dat[,2]),IRanges(start=ccds_dat$check_aa_start,end=ccds_dat$check_aa_end))
tmp=Views(Hsapiens,gr)
sequence=as.data.frame(DNAStringSet(as.character(tmp)))
ccds_dat$seq=c()
ccds_dat$trans_AA=c()
for(i in 1:nrow(ccds_dat)){
# i=4
if(ccds_dat$strand[i]==1){
# 如果是正链,直接翻译
ccds_dat[i,'seq']=sequence[i,1]
ccds_dat[i,'trans_AA']=sequence[i,1]%>% DNAStringSet(.) %>% translate()%>% as.character()
}else {
# 如果是负链,反向互补并翻译
ccds_dat[i,'seq']=sequence[i,1]%>% DNAStringSet(.) %>% reverse() %>% Biostrings::complement()%>% as.character()
ccds_dat[i,'trans_AA']=sequence[i,1]%>% DNAStringSet(.) %>% reverse() %>% Biostrings::complement() %>% translate()%>% as.character()
}
}