030-R语言20练习题
刘小泽重新写于2018.9.3
练习题来自生信技能树jimmy,http://www.bio-info-trainee.com/3409.html 再次基础上进行加工、重构,加入了自己的理解
1 安装R包
数据包: ALL, CLL, pasilla, airway
软件包:limma,DESeq2,clusterProfiler
工具包:reshape2
绘图包:ggplot2
#################################
# 1.安装R包
#################################
source("https://bioconductor.org/biocLite.R")
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
biocLite(c("ALL","CLL", "pasilla", "airway")) #数据包
biocLite(c("limma","DESeq2", "clusterProfiler")) #软件包
install.packages("reshape2") #工具包
install.packages("ggplot2") #绘图包
#另外可以检测某个包是否存在,只有不存在时才会安装
if (! require ('CLL')){
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
BiocInstaller::biocLite('CLL',ask = F, suppressUpdates = T)
}
#意思就是设定安装镜像,然后隐藏安装信息,中间过程不询问
2 了解ExpressionSet对象
CLL包中有data(sCLLex),是一个表达芯片数据对象,其中包含许多信息
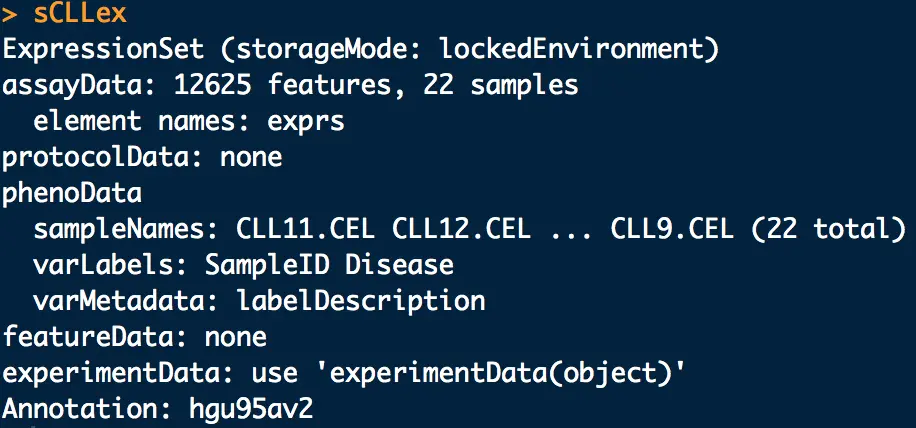
第一行的ExpressionSet就是表达矩阵,查看它使用exprs ,用dim 查看矩阵大小
#################################
# 2.了解ExpressionSet对象
#################################
suppressPackageStartupMessages(library(CLL)) #隐藏具体加载信息
#?CLL 可以看到要取出CLL数据包中的数据,需要用data函数
# CLL数据集是慢性淋巴细胞白血病(Chroniclymphocytic leukemia),它采用了Affymetrix公司的HG_U95Av2表达谱芯片(含有12625个探针组),共测量了22个样品("CLL2.CEL" "CLL3.CEL" "CLL4.CEL" "CLL5.CEL" "CLL6.CEL"等),每个样品来自一个癌症病人,所有病人根据健康状态分为两组:稳定期(Stable)组和进展期(Progressive)也称为恶化期组
## 2.1 取出数据
data("sCLLex")
#sCLLex
## 2.2 获得表达矩阵
e=exprs(sCLLex) #提取出来表达矩阵,赋给e
## 2.3 结构性探索
str(e) # 查看结构
head(e) # 查看前6行
dim(e) #查看表达矩阵大小
#结果可以看到,包含了12625个探针,22个样本
## 2.4 具体内容探索【做到完完全全了解加载的对象】
sampleNames(sCLLex) #查看样本编号
varMetadata(sCLLex) #查看所有表型变量
featureNames(sCLLex)[1:100] #查看基因芯片编码
featureNames(sCLLex) %>% unique() %>% length() #看看有没有重复,去重结果还是还是12625个
pdata=pData(sCLLex) #将样本表型信息从数据框中提取出来【取出来的是表型、样本的数据框】
group_list=as.character(pdata[,2]) #从数据框中只要表型信息
table(group_list) #统计表型信息
#看看数据质量如何
par(cex = 0.7)
n.sample=ncol(e)
if(n.sample>40) par(cex = 0.5)
cols <- rainbow(n.sample*1.2)
boxplot(e, col = cols,main="expression value",las=2)
使用str查看对象的结构,使用head查看对象的前6行(默认)
3 安装并了解hgu95av2.db包
官网:
http://www.bioconductor.org/packages/release/data/annotation/html/hgu95av2.db.html
安装
biocLite("hgu95av2.db")
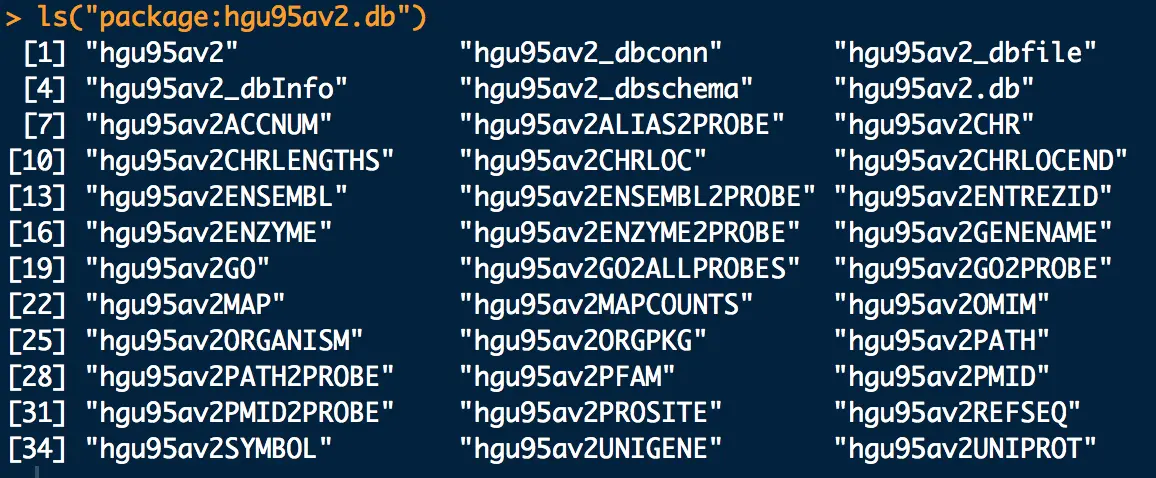
这个数据库中共有36个包,每个包都可以当成一个列表操作,可以用as.list 函数展示数据
3.1 了解hgu95av2.db 【这是一个关于hgu95av2芯片的注释包】
# Affymetrix Human Genome U95 Set annotation data
# 官方链接:https://bioconductor.org/packages/release/data/annotation/manuals/hgu95av2.db/man/hgu95av2.db.pdf
biocLite("hgu95av2.db")
library(hgu95av2.db)
ls("package:hgu95av2.db")#产生36个映射数据(探针id转为36种主流id)
capture.output(hgu95av2()) #将hgu95av2包含的具体信息输出为字符串【只用hgu95av2()可能输出,但不易整理】
#结果可以看到,每一个子集都是有keys-value构成的,就相当于list的结构。因此对于hgu95av2的操作都要使用as.list()
as.list(hgu95av2SYMBOL[1]) #变成列表进行查看第一组元素
#结果得到的就是
#$`1000_at`
#[1] "MAPK3"
#不懂hgu95av2SYMBOL是什么意思,就查找
?hgu95av2SYMBOL
#结果得到:hgu95av2SYMBOL is an R object that provides mappings between manufacturer identifiers and gene abbreviations【就是基因id与探针号的关系,这里显示的就是MAPK3这个基因对应的探针id是1000_at】
3.2 探索hgu95av2CHR:探针id与染色体编号对应的关系
C = hgu95av2CHR #首先明白CHR它的数据结构,左边一列是探针id,右边一列是chr id
Llength(C) #计算左侧的数量,同理Rlength()
Rkeys(C) #查看右侧的名称,同理Lkeys()
Rkeys(C) =c("6","8") #只保留chr6、chr8的数据
table(toTable(C)[,2]) #统计chr6上有668个探针,chr8上有402个探针
3.3 探索hgu95av2SYMBOL:探针id与基因名(缩写)的关系
3.3.1 先对探针id进行简单的探索:【输出探针】
s = hgu95av2SYMBOL
summary(s) #一个函数搞定左右两边的各种统计【包括总数、过滤后的】
#比如:左边探针总数是12625,能匹配上的是11430个;
#右边:基因名的总数是30071,实际上只有8585种
#【【那么,为什么基因名过滤前后差别如此大呢?】】
#[下面]目的:对参考注释进行过滤,去掉没有map的探针
mapped_probes <- mappedkeys(s) #手动过滤,然后进行统计
count.mappedkeys(s) #统计:与summary结果相符,有11430个探针有对应基因名称的【剩余1165没有名称】
问题来了,剩下没有匹配上1165个探针的id都是什么呢?换句话说,找到没有在hgu95av2SYMBOL中有对应SYMBOL的探针
#先自定义一个函数:表示找到不在某个范围内的,返回值是T/F
'%!in%' <- function(x,y)!('%in%'(x,y))
Lkeys(s)[Lkeys(s)%!in% mapped_probes]
3.3.2 再对基因名进行探索【匹配、列表、转数据框、查特定基因、基因数】
ss = as.list(s[mapped_probes]) #将匹配上的基因输出为列表
sst=toTable(s[mapped_probes]) #toTable把list转换成数据框 【后面会经常用到sst】
colnames(sst) #看一下sst的列名【相当于summary(s)中的Lkeyname=probe_id、Rkeyname=symbol】
sst[grep("^TP53$",sst$symbol),] #查找TP53基因对应的探针id
unique(sst$symbol) %>% length() #查看总共基因数(注意unique的使用,为了避免重复基因出现)
table(sst$symbol) %>% sort() %>% tail() #table将基因名字符量化为数字,sort从小到大排序,tail取最大6个
table(sst$symbol) %>%table() #table函数统计不同探针数量对应的基因数
#【【为什么有5个基因会分别有8个探针,而大部分6555个基因只对应一个探针?】】
为什么有5个基因会分别有8个探针,而大部分6555个基因只对应一个探针?
A:不管是Agilent芯片,还是Affymetrix芯片,上面设计的探针都非常短。最长的如Agilent芯片上的探针,往往都是60bp,但是往往一个基因的长度都好几Kb。因此一般多个探针对应一个基因,取最大表达值探针来作为基因的表达量
4 过滤、整合表达矩阵
4.1 过滤
#[下面]目的:对表达矩阵进行过滤,去掉没有map的探针
# table(rownames(e)%in%sst$probe_id) #找到sCLLex表达矩阵(e)在hgu95av2.db包中没有交叉的探针
# %>%是管道符号,相当于linux的“|”; %in%表示两者求交集
e1 = e[rownames(e)%in%mapped_probes,] #对原始表达矩阵过滤
# e2 = e[match(rownames(e), mapped_probes, nomatch = 0),] #使用match过滤
4.2 整合
#[下面]目的:一个基因对应一个探针
#现状分析:多个探针对应一个基因的情况存在
#解决途径:只保留在所有样本里面平均表达量最大的那个探针【一般采用均值即可,当然也可用最大值】
#(之前得到的sst矩阵来的时候就是根据mapped_probes得到的,也就是过滤好的,因此可以直接用作index)
maxp = by(e1,sst$symbol,function(x) rownames(x)[which.max(rowMeans(x))]) #矩阵用by,向量用tapply
uniprobes = as.character(maxp) #获得每个基因独特的探针
efilt=e[rownames(e)%in%uniprobes,] #完成表达矩阵过滤
rownames(efilt)=sst[match(rownames(efilt),sst$probe_id),2]
4.3 重塑【reshape2:矩阵=》数据框】
head(efilt) #可以看到现在还是一个矩阵的样子,行为基因名,列为样本名
#我们想把这个矩阵变成标准的tidy data,三列:第一列基因名,第二列样本名,第三列表达量
library(reshape2)
m_efilt = melt(efilt) #先将原来矩阵“融化
colnames(m_efilt)=c('symbol','sample','value') #重新命名三列
m_efilt$group=rep(group_list,each=nrow(efilt)) #最后再加一列表型信息(就是刚得到表达矩阵时提取的group_list),每一个样本有8585行【nrow(efilt)统计得到】,我们这里重塑的数据框是把所有样本按次序堆叠起来,因此每个样本的表型应该将group_list中对应的表型重复nrow(efilt)这些次
#例如,第一个样本是CLL11.CEL,它的表型是progres。现在我们把CLL11.CEL放到重塑的sample列,应该就有8585行都是CLL11.CEL,然后再有8585个样本CLL12.CEL。因此CLL11.CEL对应的表型信息也就应该是8585个,也就是融化前矩阵的行数
#融化前后做个对比就看出来了
head(efilt)
head(m_efilt)
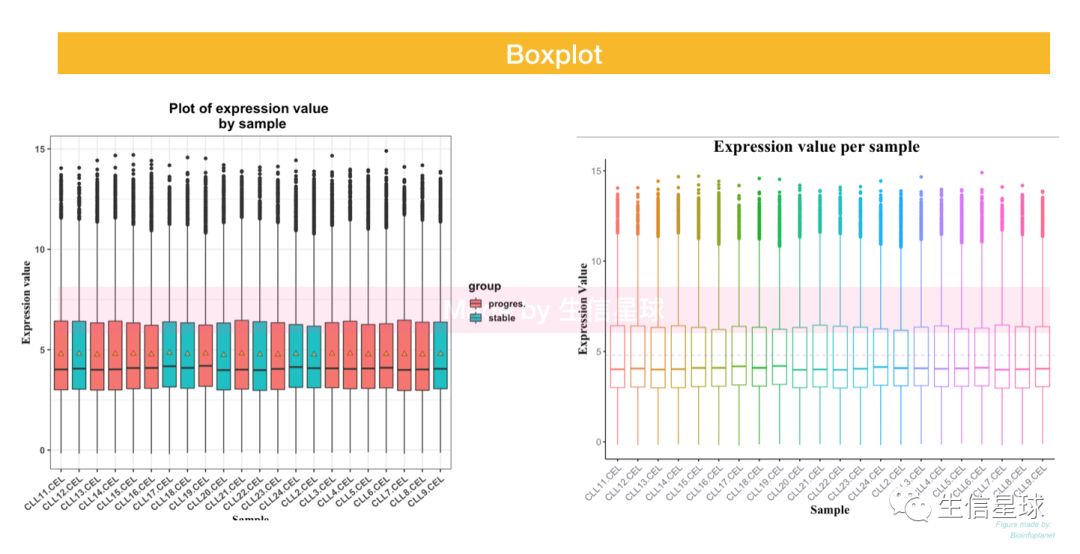
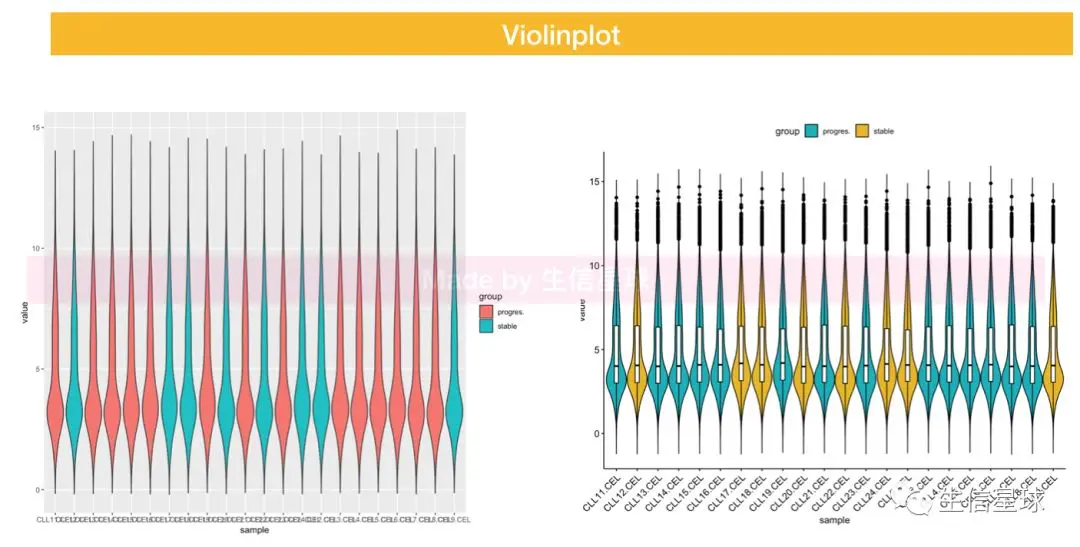
5 画图探索
作出样本和基因表达量之间的关系图,主要基于ggplot 每种图都做了两种版本,一个初始版,一个调整版
5.1 boxplot
5.2 violin plot
5.3 Histogram
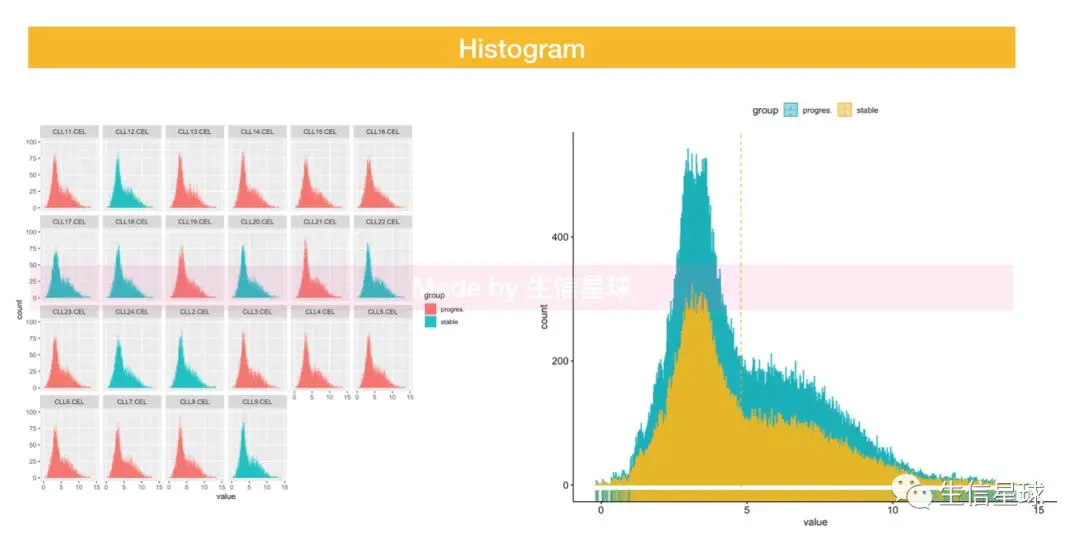
5.4 Density
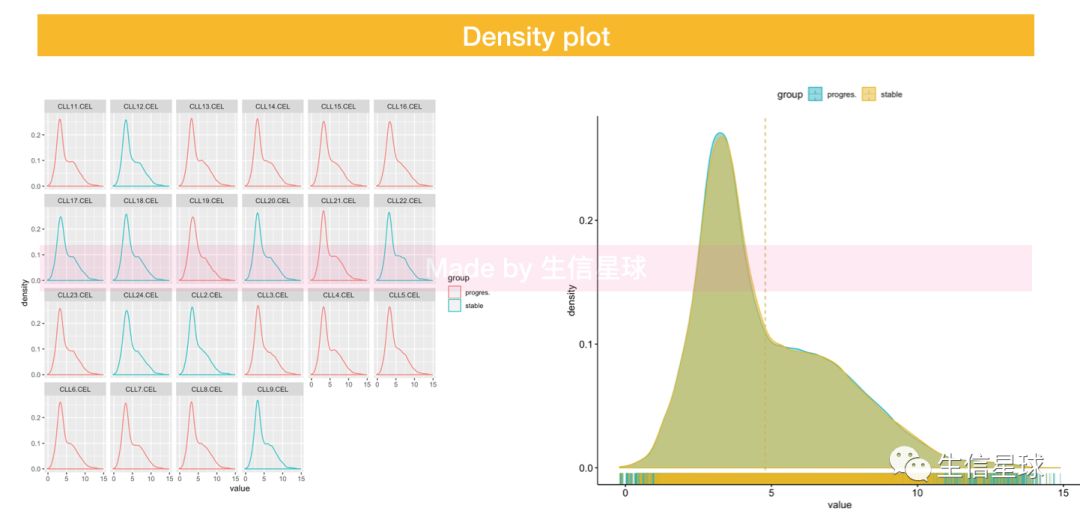
6 做一些统计
mean,median,max,min,sd,var,mad,t检验
6.1 利用apply函数进行统计
他需要矩阵,也就是之前得到的efilt,按行进行统计即可
e_mean = tail(sort(apply(efilt,1,mean)),30)
e_median = tail(sort(apply(efilt,1,median)), 30)
e_max <- tail(sort(apply(efilt,1,max)),30)
e_min <- tail(sort(apply(efilt,1,min)),30)
e_sd <- tail(sort(apply(efilt,1,sd)),30)
e_var <- tail(sort(apply(efilt,1,var)),30)
e_mad <- tail(sort(apply(efilt,1,mad)),30) #绝对中位差来估计方差,先计算出数据与它们的中位数之间的偏差,然后这些偏差的绝对值的中位数就是mad
6.2 看表达量top30基因之间的重合部分
用UpSetR包结合之前做的top30基因各种统计,适用于样本数量大于5的情况
其实我们平常做的韦恩图也是这个意思,找交集,但是韦恩图样本数量一般都会控制在
install.packages("UpSetR")
library("UpSetR")
#先找出7个统计量的共同基因名
e_all <- c(names(e_mean),names(e_median),names(e_max),names(e_min),
names(e_sd),names(e_var),names(e_mad)) %>% unique()
#分别将7个统计量关于共同基因名的统计值
edat=data.frame(e_all,
e_mean=ifelse(e_all %in% names(e_mean) ,1,0),
e_median=ifelse(e_all %in% names(e_median) ,1,0),
e_max=ifelse(e_all %in% names(e_max) ,1,0),
e_min=ifelse(e_all %in% names(e_min) ,1,0),
e_sd=ifelse(e_all %in% names(e_sd) ,1,0),
e_var=ifelse(e_all %in% names(e_var) ,1,0),
e_mad=ifelse(e_all %in% names(e_mad) ,1,0)
)
upset(edat,nsets = 7,sets.bar.color = "#56B4E9")
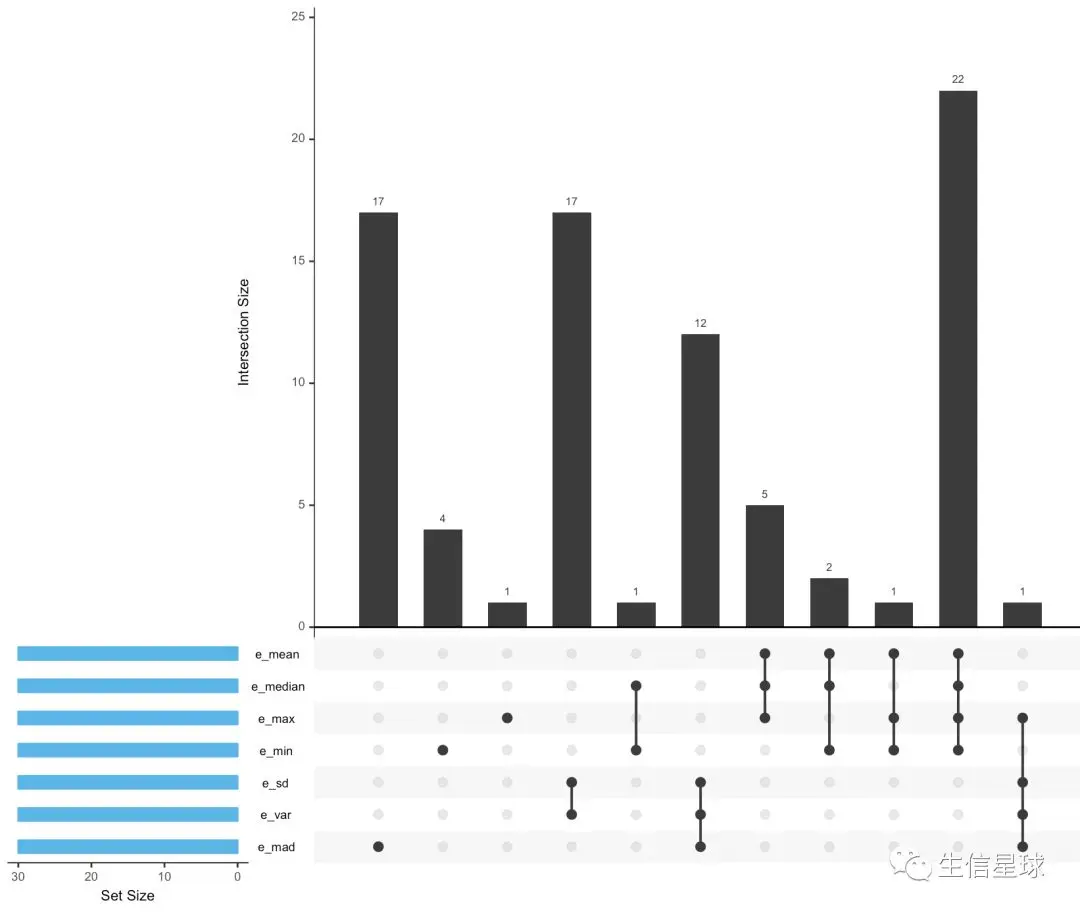
6.3 批量T检验——为了得到pvalue【后续分析重点】
有了pvale就能有padj值;另外还需要对照、处理两组均值,这样就能有log2FC
gl=as.factor(group_list) #将最初得到的的表型数据因子化
group1 = which(group_list == levels(gl)[1])
#levels(group_list)[1]返回第一个因子progres,从group_list中选出progres的元素,用which来获取他们所在的位置【目的是为下面分别得到两种表型的样本作准备】
group2 = which(group_list == levels(gl)[2]) #返回第二个因子stable
et1 = e[, group1] #将表型为progres的样本选出来,因为这里是要求t值,可以命名为e矩阵的t值,即et
et2 = e[, group2] #将表型为stable的样本选出来
et = cbind(et1, et2) #按列合并
pvals = apply(e, 1, function(x){
t.test(as.numeric(x)~group_list)$p.value # 多组样本的t检验
})
p.adj = p.adjust(pvals, method = "BH") #多重比较时校正p值
eavg_1 = rowMeans(et1) #progres是对照组
eavg_2 = rowMeans(et2) #stable是使用药物处理后的——处理组
log2FC = eavg_2-eavg_1
DEG_t.test = cbind(eavg_1, eavg_2, log2FC, pvals, p.adj)
DEG_t.test=DEG_t.test[order(DEG_t.test[,4]),] #从小到大排序
DEG_t.test=as.data.frame(DEG_t.test)
head(DEG_t.test)
一般来讲,下游分析使用的差异表达矩阵应该是log2后的结果,它的计算公式是log2FC = log2 (mean(处理组/对照组))
这里为什么可以之间相减? 芯片数据的特点:小样本和大变量,因此数据分布呈偏态、标准差大。而对数转换能使上调、下调的基因连续分布在0的周围,更加符合正态分布,同时对数转换可以使荧光信号强度的标准差减少,方便下游分析 因此我们一直用的e也就是exprs(sCLLex)得到的表达矩阵是log2之后的 我们得到的eavg_2 = log2(mean处理组),eavg_1 = log2(mean处理组), 根据公式就可以算出log2(a/b) = log2(a) - log2(b)
7 做一些分析 表达量、聚类、PCA、火山图
7.1 按mad指标选表达量前30(top30)的基因,做热图可视化
做热图前需要将矩阵数据中心化+标准化【目的为了向数据中心靠拢,减小数据之间的差别】
中心化:数据减去均值后得到的; 标准化则是在中心化后的数据基础上再除以数据的标准差
top30_gene=names(e_mad)
top30_matrix=efilt[top30_gene,] #得到top30的表达量矩阵
top30_matrix=t(scale(t(top30_matrix)))
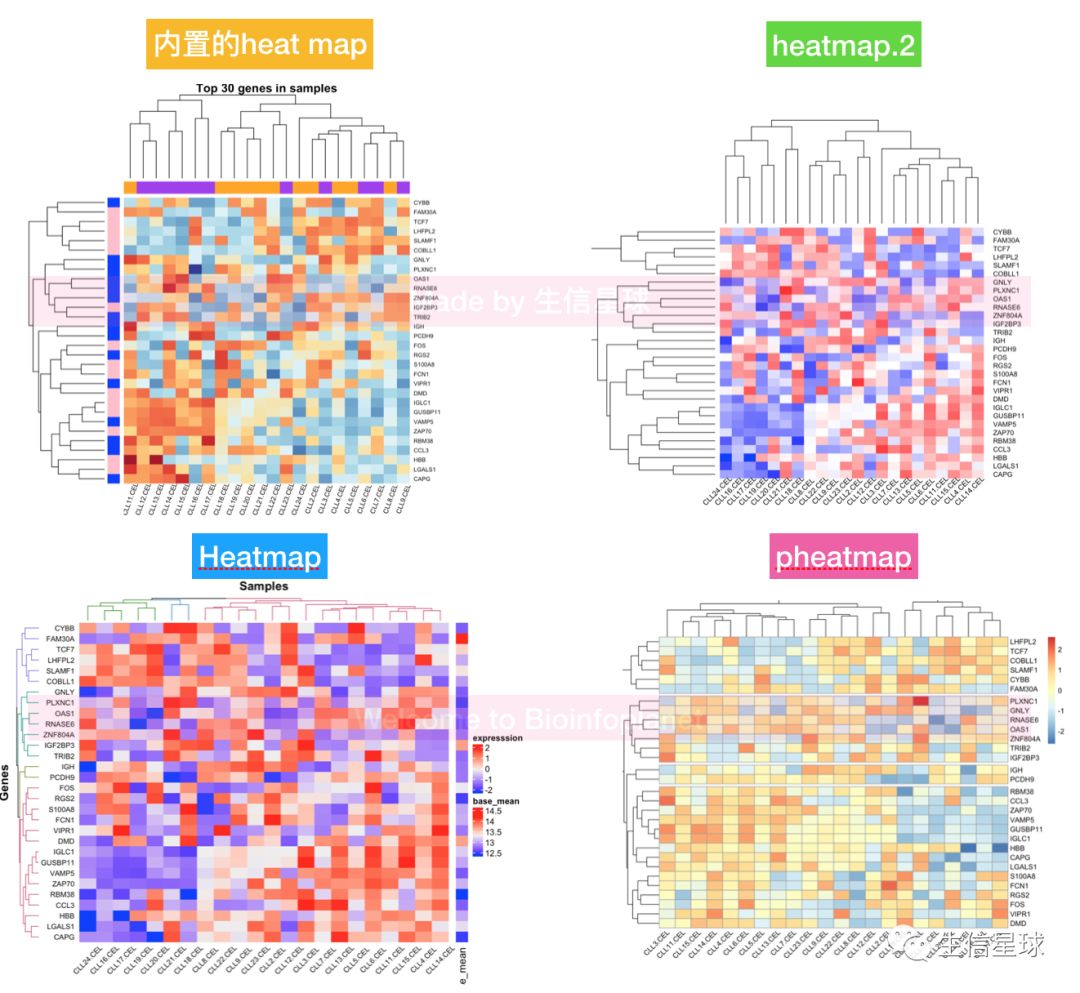
7.2 聚类分析图
过滤后的样本聚类
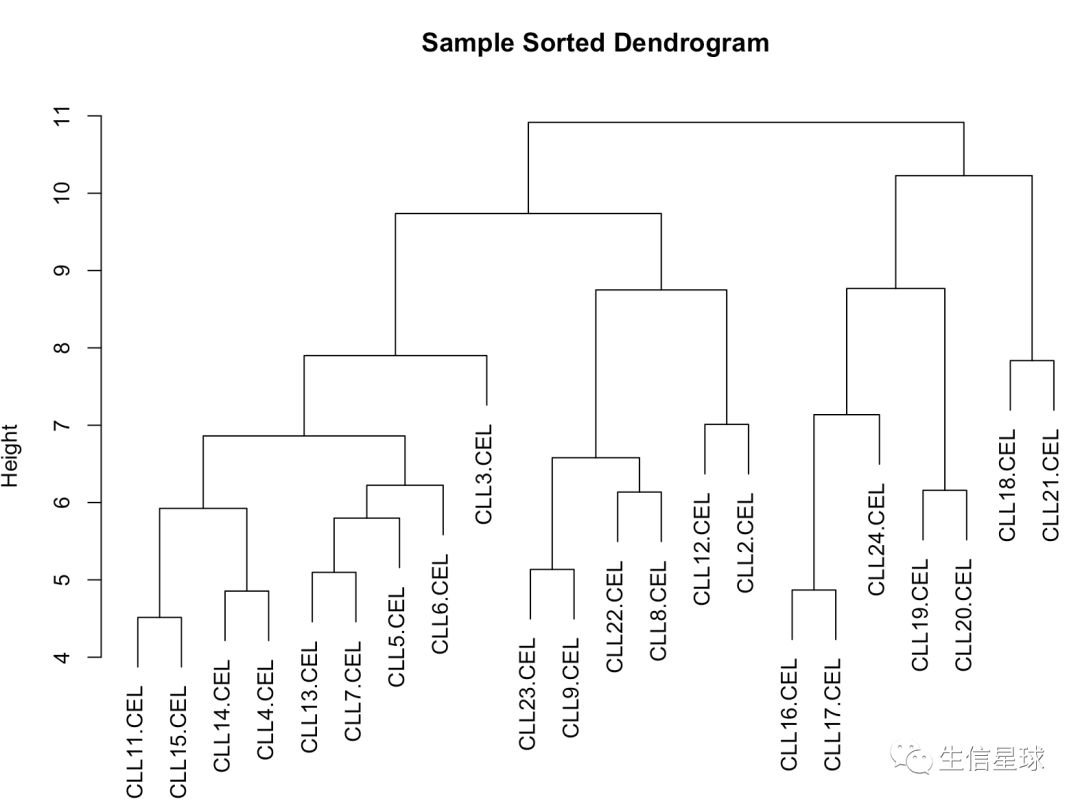
过滤后的基因聚类

使用factoextra包
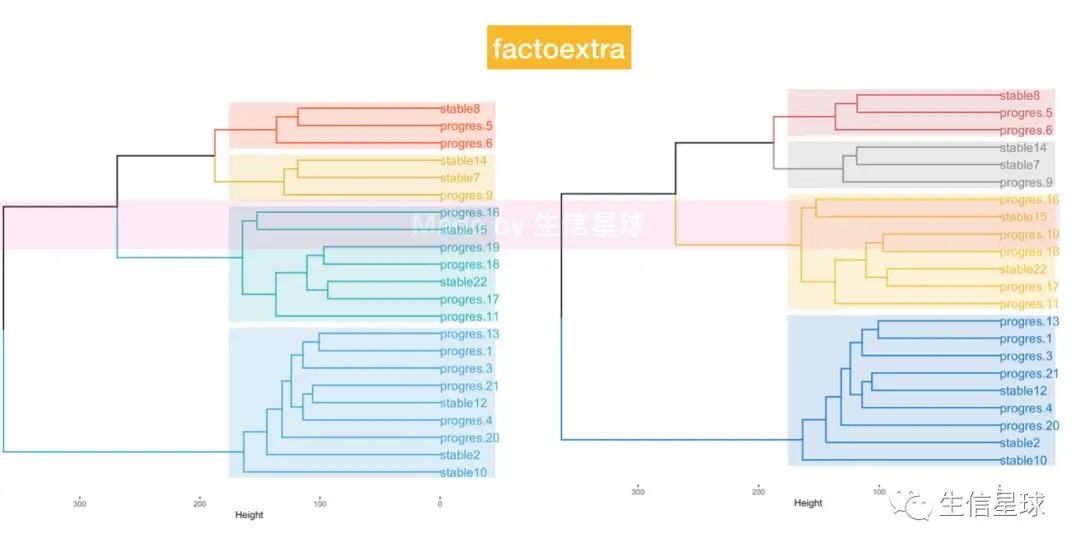
如果看到聚类分析的结果枝干太长,那么就要换种聚类方法
7.3 PCA分析
【目的:简化变量的个数】本质是降维,本来应该有22个变量,现在我们变成了22个主成分,一般前面的几个主成分就能解释所有的数据。解释:https://wenku.baidu.com/view/c22d1539a31614791711cc7931b765ce05087a6f.html http://www.bio-info-trainee.com/1232.html
7.3.1 使用一键式ggfortify + prcomp
关于ggfortify的使用:https://wenku.baidu.com/view/e5dc63fb763231126fdb1100.html 最大的优点:一行代码,出ggplot风格的图,不用费时调整,提高效率

结果中:不同颜色代笔不同分组; 坐标轴:能最大反映样本差异性的两个成分(PC1、PC2) 百分数:成分贡献率; 坐标轴刻度:没实际意义(代表相对距离)
7.3.2 使用fviz_pc
a_ind包
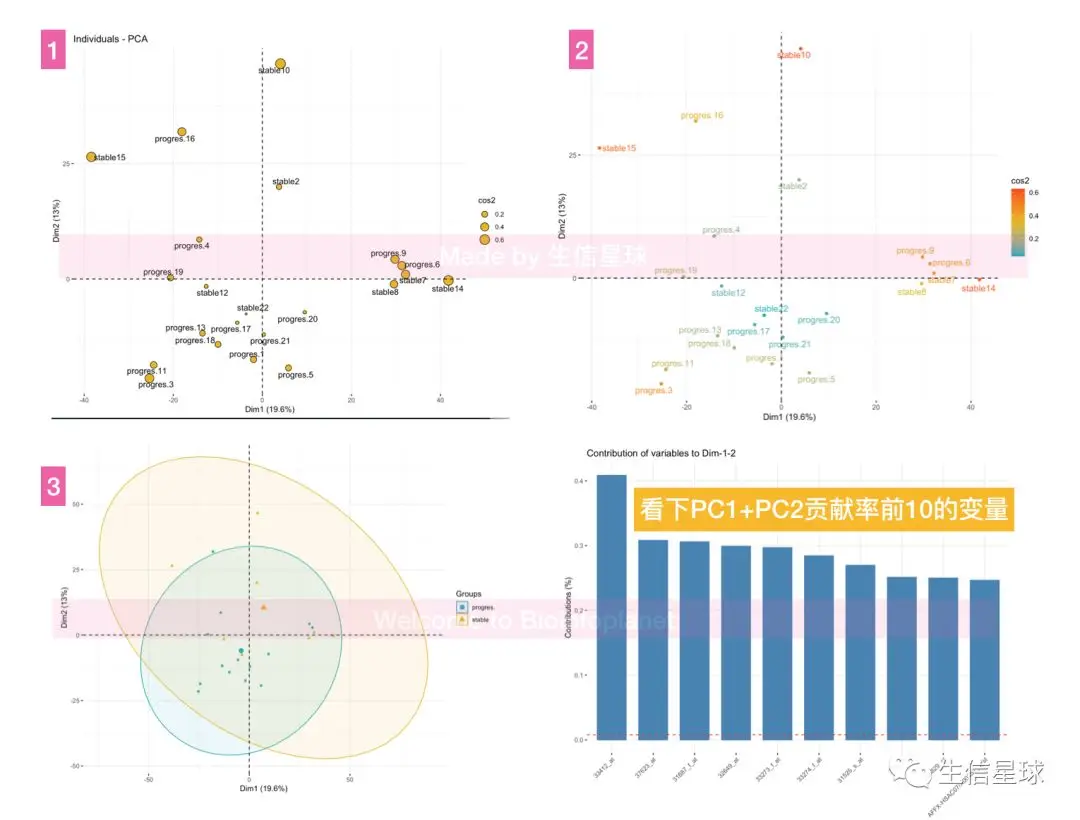
7.4 差异分析火山图【也就是logFC加上-log10(Pvals)的散点图】
首先构建差异分析矩阵
suppressMessages(library(limma)) #加载包不显示啰嗦的信息
关于limma:
limma是基于R和Bioconductor平台的分析芯片数据的综合软件包,该包功能齐全、教程完善、使用率极高,几乎成为了芯片数据处理流程的代名词; 本质就是对表达量矩阵做一个归一化,然后利用理想的统计分布检验函数来计算差异的显著性; imma的核心函数是lmFit和eBayes, 前者是用于线性拟合,后者根据前者的拟合结果进行统计推断; lmFit至少需要两个输入,一个是表达矩阵,一个是分组对象; 表达矩阵必须是matrix类数据结构,每一列都是存放一个样本,每一行是一个探针信息或者是注释后的基因名
关于比较矩阵: 参考https://github.com/bioconductor-china/basic/blob/master/makeContrasts.md
7.4.1 构建一个非差异比较矩阵【只有一组处理和对照,所以可以不用比较矩阵】
design1=model.matrix(~factor(group_list))
colnames(design1)=levels(factor(group_list))
rownames(design1)=colnames(e)
fit1 = lmFit(e,design1)
fit1=eBayes(fit1)
options(digits = 3) #设置结果的小数位数为3
mtx1 = topTable(fit1,coef=2,adjust='BH',n=Inf) #coef要么必须等于2, 要么是个字符串;关于adjust的设置,说明书中13.1章有描述,BH是最流行的设置
# topTable 默认显示前10个基因的统计数据;使用选项n可以设置,n=Inf就是不设上限,全部输出
DEG_mtx1 = na.omit(mtx1) #去除缺失值
head(DEG_mtx1)
7.4.2 构建差异比较矩阵
#【当有多组处理、对照时就发挥作用了,例如两个细胞系A、B,每个细胞系都有处理、对照组】
##先分组##
design2=model.matrix(~0+factor(group_list))
colnames(design2)=levels(factor(group_list))
rownames(design2)=colnames(e)
fit2=lmFit(e,design2)
##后比较##
contrast.matrix<-makeContrasts(paste0(unique(group_list),collapse = "-"),levels = design2) #注意写法:处理和对照之间用-隔开
#假如是A、B两组处理、对照的情况,makeContrasts(A_trt-A_con,B_trt-B_con),levels = design2)
#另外,paste0相当于paste(..., sep = "", collapse = ) 将...对象拼起来,并且sep指定对象们无缝拼接,得到的结果用collapse分隔
# 如果paste(unique(group_list),sep = "-") =》"progres." "stable"
# 如果paste0(unique(group_list),collapse = "-") =》"progres.-stable" 【一个整体,只是中间有分隔】
fit2=contrasts.fit(fit2, contrast.matrix)
fit2=eBayes(fit2)
##得矩阵##
mtx2=topTable(fit2, coef=1, n=Inf) #如果是多组,还是上面的例子:如果需要第一组A的差异基因,就用coef = 1;如果需要第二组B的差异基因,就用coef = 2;但是如果是多组比较,结果容易什么差异基因都没有,要注意批次效应!使用h-cluster来辨识批次效应,用combat来校正
DEG_mtx2= na.omit(mtx2)
#write.csv(nrDEG2,"limma_notrend.results.csv",quote = F)
head(DEG_mtx2)
#就结果来看,DEG_mtx1和DEG_mtx2是一样的
#########################################################
7.4.3 准备火山图
画火山图第一步,设定阈值,选出UP、DOWN、NOT表达基因
DEG=DEG_mtx2
logFC_cutoff <- with(DEG,mean(abs(logFC)) + 2*sd(abs(logFC)) ) # with()函数适用于当同名变量出现多次,避免程序定位错误的情况,with()的括号内外,信息是完全隔绝的。因为需要使用DEG中的logFC,但logFC只在DEG中出现,当然可以单独用DEG$logFC选出来设为全局变量,但是感觉有点“兴师动众”,此时用with就是最好的情况
关于阈值的设置:一般情况都是设为2,但具体背景不同还是应该设置不同的阈值。根据网站https://www.bmj.com/about-bmj/resources-readers/publications/statistics-square-one/2-mean-and-standard-deviation 的介绍。mean+2SD可以反映95%以上的观测值,这是比较可信的,如果再严格一点,设为mean+3SD,就可以反映97%以上的观测
DEG$result = as.factor(ifelse(DEG$P.Value < 0.05 & abs(DEG$logFC) > logFC_cutoff,
ifelse(DEG$logFC > logFC_cutoff ,'UP','DOWN'),'NOT')
)
#这是两个ifelse判断嵌套。先了解ifelse的结构,ifelse(条件,yes,no),如果满足条件,那么返回yes/或者执行yes所处的下一个命令;反之返回no
#这里外层的ifelse中DEG$P.Value < 0.05 & abs(DEG$logFC) > logFC_cutoff是判断条件,这个就是看p值和logFC是不是达到了他们设定的阈值【p是0.05,logFC是logFC_cutoff】,如果达到了就进行下一个ifelse,达不到就返回NOT;
#第二层ifelse也是上来一个条件:DEG$logFC > logFC_cutoff,如果达到了,就返回UP即上调基因,达不到就是下调DOWN
# 最后将判断结果转位因子型,得到DOWN、UP、NOT的三种因子
画火山图第二步,设定图形的标题
this_tile <- paste0('Cutoff for logFC is ',round(logFC_cutoff,3), #round保留小数位数
'\nThe number of up gene is ',nrow(DEG[DEG$result =='UP',]) ,
'\nThe number of down gene is ',nrow(DEG[DEG$result =='DOWN',])
)
最后才开始画火山图
library(ggplot2)
#注意DEG的列名不同的分析软件可能命名不同,比如p值,有的是P.value,有的是P_value
#选出p值并且进行对数转换
#其实火山图就是不同颜色的散点图,只不过低于阈值的居多,并且设为深颜色,所以看着像火山喷发
ggplot(data=DEG, aes(x=logFC, y=-log10(P.Value), color=result)) +
geom_point(alpha=0.4, size=1.75) +
theme_set(theme_set(theme_bw(base_size=20)))+
xlab("log2 fold change") + ylab("-log10 p-value") +
ggtitle( this_tile ) + theme(plot.title = element_text(size=15,hjust = 0.5))+
scale_colour_manual(values = c('blue','black','red')) ## 这里要注意和之前设置的result三个因子相对应,DOWN就设为blue,NOT就设为black