246-资源分享——常用的pipeline这里都有
简介
最近看到一个不错的网站nf-core,其中分享了各个NGS组学分析流程,关于它的历史:
The nf-core project came about at the start of 2018. Phil Ewels (@ewels) was the head of the development facility at NGI Stockholm (National Genomics Infrastructure), part of SciLifeLab in Sweden.
To try to open up the effort into a truly collaborative project, nf-core was created and all relevant pipelines moved to this new GitHub Organisation.
All nf-core pipelines are written in Nextflow and so inherit the ability to be executed on most computational infrastructures, as well as having native support for container technologies such as Docker and Singularity.
足以见得项目的庞大
这个项目还发表在了:Nat Biotechnology (2020); doi: 10.1038/s41587-020-0439-x The nf-core framework for community-curated bioinformatics pipelines
49种pipeline
它的所有流程都是用Nextflow穿起来的
已经可以使用的流程有30个,主要包括了(有好多甚至都没听说过):
- DIA proteomics mass spectrometry measurements
- CLIP sequencing analysis
- ProteoGenomics data analysis.
- ChIP-seq
- ancient DNA analysis pipeline
- scRNA
- Methylation (Bisulfite-Sequencing) analysis
- Dual RNA-seq data
- metagenomes
- 16S rRNA amplicon sequencing analysis
- detect germline or somatic variants
- CAGE-sequencing analysis
- RNAseq
- Simple bacterial assembly & annotation
- epitope prediction and annotation
- Proteomics label-free quantification (LFQ)
- Identify and quantify MHC eluted peptides from mass spectrometry
- HiC
- Precision HLA typing
- ATAC-seq
- RNAseq for detection gene-fusions
- low-frequency variant calling for viral samples
- Image Mass Cytometry analysis
- SLAMSeq
- Coprolite host Identification
- B cell repertoire sequencing analysis
- Small-RNA sequencing
- Google’s DeepVariant variant caller

当然还有很多如smartseq2、GWAS、circRNA 等分析流程在开发中
参与机构目测也有几十个
https://nf-co.re/community#organisations

以ATAC-seq为例
https://nf-co.re/atacseq/1.2.1/output

主体流程包括:
- Raw read QC (FastQC)
- Adapter trimming (Trim Galore!)
- Alignment (BWA)
- Mark duplicates (picard)
- Filtering to remove(samtools)
- Alignment-level QC and estimation of library complexity (picard, Preseq)
- Create normalised bigWig files scaled to 1 million mapped reads (BEDTools, bedGraphToBigWig)
- Generate gene-body meta-profile from bigWig files (deepTools)
- Calculate genome-wide enrichment (deepTools)
- Call broad/narrow peaks (MACS2)
- Annotate peaks relative to gene features (HOMER)
- Create consensus peakset across all samples and create tabular file to aid in the filtering of the data (BEDTools)
- Count reads in consensus peaks (featureCounts) Differential accessibility analysis, PCA and clustering (R, DESeq2)
- Create IGV session file containing bigWig tracks, peaks and differential sites for data visualisation (IGV).
- Present QC for raw read, alignment, peak-calling and differential accessibility results (ataqv, MultiQC, R)
当然,它在比对后进行了很多过滤(Filtering to remove)的操作,可以借鉴:
- reads mapping to mitochondrial DNA (SAMtools)
- reads mapping to blacklisted regions (SAMtools, BEDTools)
- reads that are marked as duplicates (SAMtools)
- reads that arent marked as primary alignments (SAMtools)
- reads that are unmapped (SAMtools)
- reads that map to multiple locations (SAMtools)
- reads containing > 4 mismatches (BAMTools)
- reads that are soft-clipped (BAMTools)
- reads that have an insert size > 2kb (BAMTools; paired-end only)
- reads that map to different chromosomes (Pysam; paired-end only)
- reads that arent in FR orientation (Pysam; paired-end only)
- reads where only one read of the pair fails the above criteria (Pysam; paired-end only)
结果文件
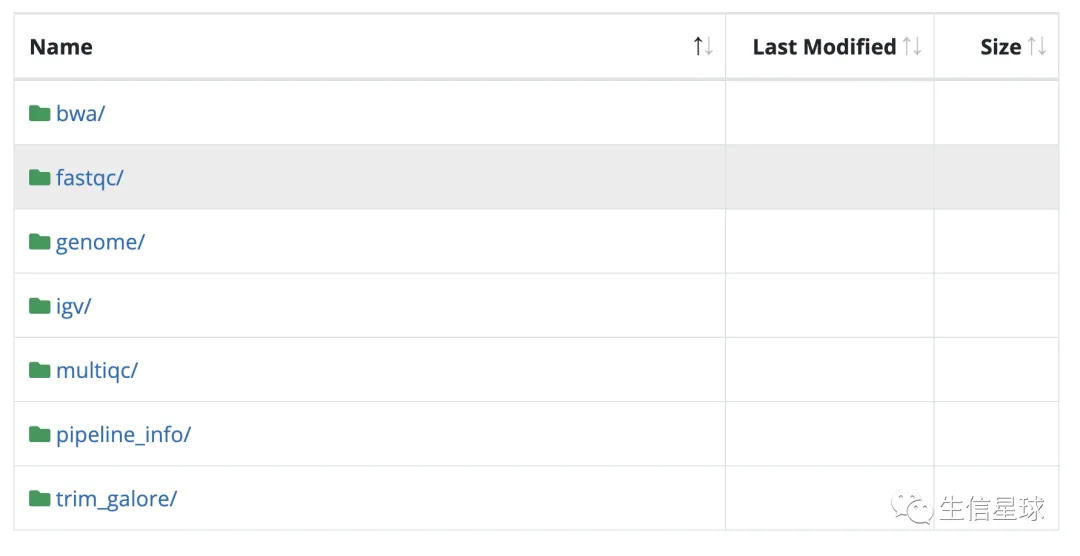
最后也会用multiqc给出一个报表
https://nf-co.re/atacseq/1.2.1/output
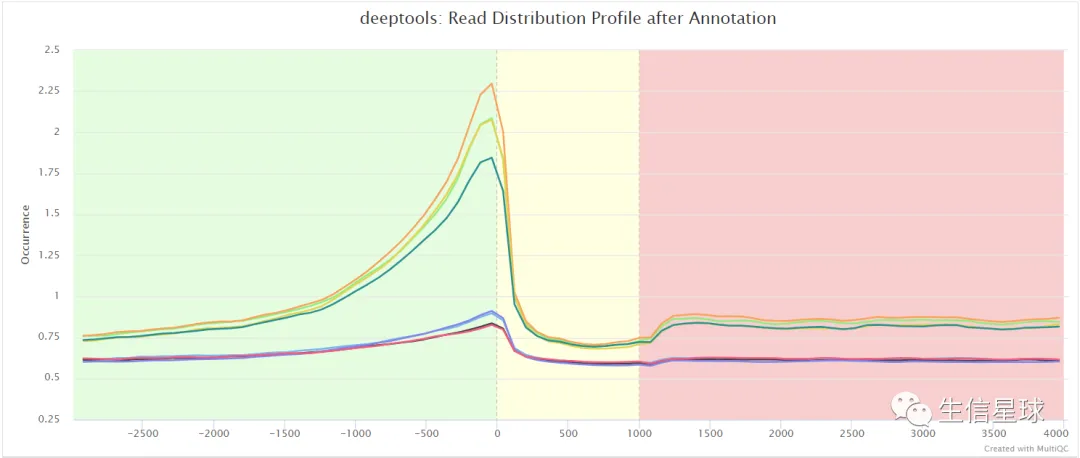
比如信号强度:
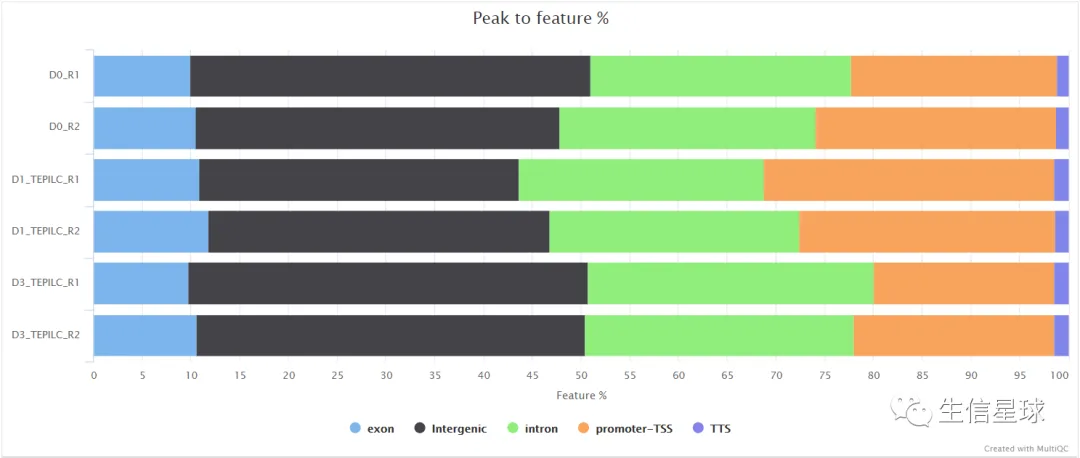
比如peaks的注释:
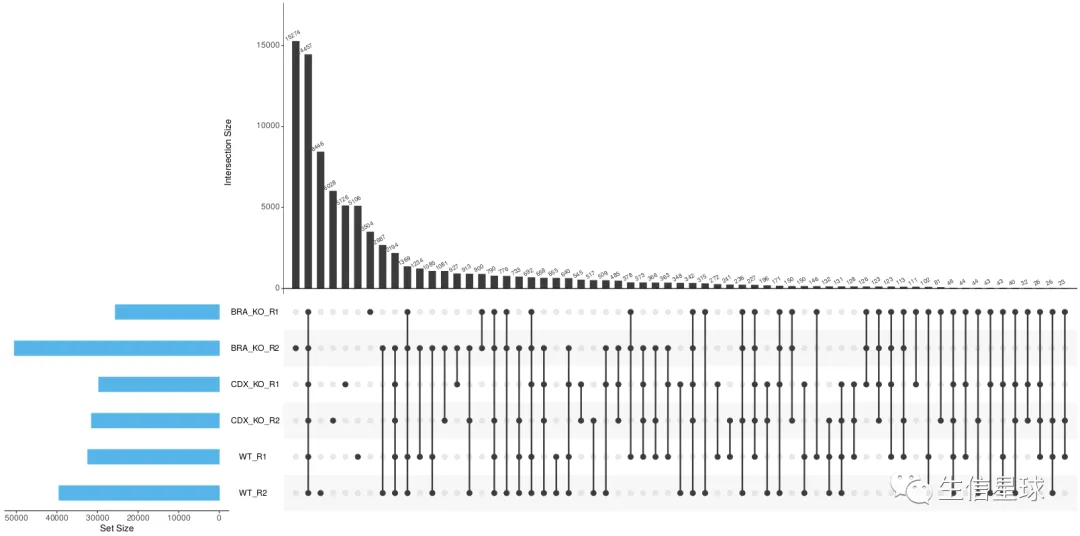
比如peaks的交集: