249-差异分析之前到底要不要进行基因过滤?
刘小泽写于2021.6.18
前言
我们在分析RNA-seq的差异分析时,总会对原始的表达矩阵进行过滤,当然过滤方式多种多样,也各有各的道理。但是今天我被问到一个问题:你为什么要过滤?过滤有什么好处呢?
对啊,我们习惯了常规的操作,看别人进行了过滤,我们也进行过滤。但是为什么呢?
原因
一:生物学角度
我们一般会把在所有样本中都不表达 的基因过滤掉,这也许是最简单的方式了。
这些在所有样本中表达量为0的基因,虽然有可能是由于测序原因或者本身表达量就很低,而得不到有效数值,但是我们还是选择相信它在生物变化过程中作用比较小,没理由放着成千上万有差异的基因不管,而在这些全是0表达量的基因上浪费时间。
所以,为了更好解释生物问题,我们要过滤
二:统计学角度
不进行过滤,无疑会在差异分析中增加背景基因数量,而差异分析实际上也是基于数据分布完成的统计检验(只不过涉及的模型和检验方法更加复杂),如果引入太多的”背景噪音“,那么检验的灵敏度和准确度都会降低。
这里,之前有人还发了1区9分的文章:Independent filtering increases detection power for high-throughput experiments (Proc Natl Acad Sci U S A. 2010 May 25;107(21):9546-51.)
- Variable-by-variable statistical testing is often used to select variables whose behavior differs across conditions. Such an approach requires adjustment for multiple testing.
- A two-stage approach that first filters variables by a criterion independent of the test statistic, and then only tests variables which pass the filter, can provide higher power.
再比如这篇估计大家都看过的文章:RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR (Version 3. F1000Res. 2016; 5: ISCB Comm J-1408.) 也提到:
- Genes/transcripts having read count within 0-10 rage are generally considered as artifacts or ‘noise’
- From a statistical point of view, removing low count genes allows the mean-variance relationship in the data to be estimated with greater reliability
当然,这个0-10是否太宽泛我们先不管,他也认为表达量太低会是噪音污染
所以,为了更好解释统计问题,我们要过滤
再看一个biostar的讨论
意思就是:不仅要过滤,还有变着花样地过滤(比如有人专门开发了一个包去做这件事)

如果就是不过滤会怎样?
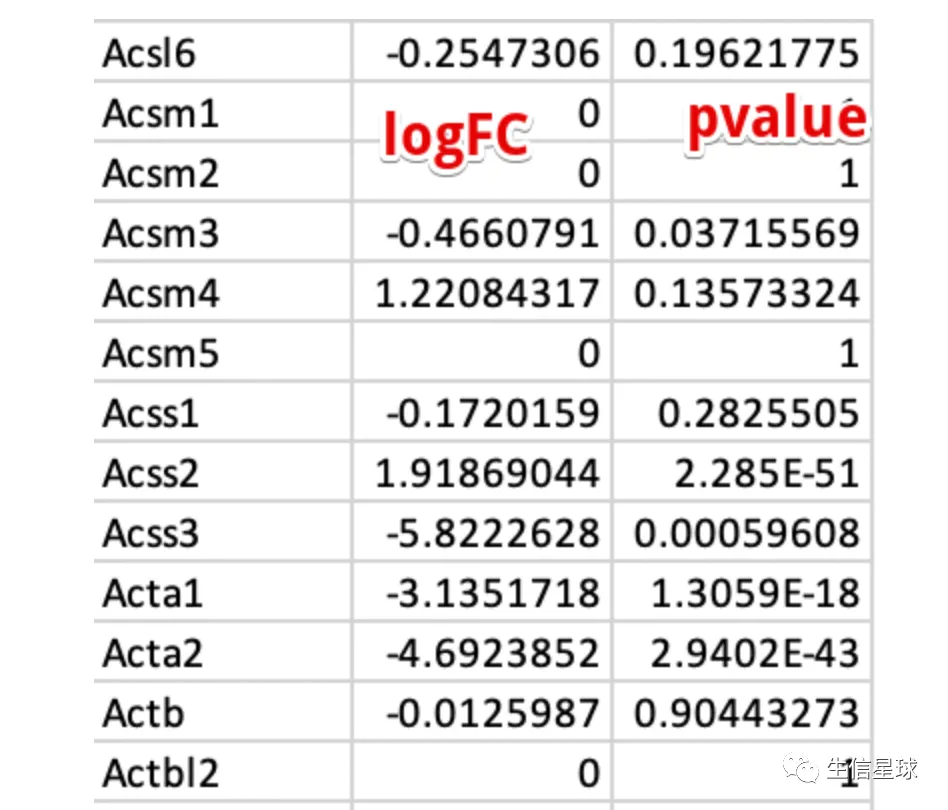
给出一个例子,我对原始的表达矩阵不进行任何的过滤,给到edgeR
看到其中大量的logFC = 0 就是由于该基因在所有样本中都不表达的结果
如果要过滤,有什么办法吗?
我们拿GTEX中一个样本数量比较少的组织 ovary来测试
首先获取数据
library(TCGAbiolinks)
data <-TCGAquery_recount2(project="GTEX", tissue="ovary")
# tissuesGTEx <- c("adipose_tissue", "adrenal_gland", "bladder",
# "blood", "blood_vessel", "bone_marrow", "brain", "breast",
# "cervix_uteri", "colon", "esophagus", "fallopian_tube",
# "heart", "kidney", "liver", "lung", "muscle", "nerve",
# "ovary", "pancreas", "pituitary", "prostate", "salivary_gland",
# "skin", "small_intestine", "spleen", "stomach", "testis",
# "thyroid", "uterus", "vagina")
x <- data[[1]]
> x
class: RangedSummarizedExperiment
dim: 58037 108
metadata(0):
assays(1): counts
rownames(58037): ENSG00000000003.14 ENSG00000000005.5 ...
ENSG00000283698.1 ENSG00000283699.1
rowData names(3): gene_id bp_length symbol
colnames(108): SRR1389909 SRR1454460 ... SRR1086212
SRR1431514
colData names(82): project sample ... title
characteristics
所有样本都不表达的基因
108个样本中,有1164个基因都不表达
library(SummarizedExperiment)
> table(rowSums(assay(data[[1]])==0)==ncol(x))
FALSE TRUE
56873 1164
使用edgeR的filterByExpr进行过滤
这个函数的定位是: an automatic way to filter genes, while keeping as many genes as possible with worthwhile counts 它的方法是:keeps genes that have at least
min.countreads in a worthwhile number of samples. More precisely, filtering keeps genes that have count-per-million (CPM) above_k_in_n_samples, where_k_is determined bymin.countand by the sample library sizes and_n_is chosen according to the minimum group sample size
也就是说,它不是对原始表达矩阵进行过滤,而是对构建完差异比较矩阵后,正式差异分析之前,进行的过滤
# 需要一个样本信息group_list(指定control和treat),注意它需要是因子型
# 我这里就简单粗暴指定前一半是treat,后一半是control
group_list=factor(rep(c("treat","control"),each=ncol(x)/2),levels = c("control","treat"))
e=DGEList(counts=assay(x),group=factor(group_list))
keep.exprs <- filterByExpr(e, min.count=10)
filt1 <- x[keep.exprs,]
# 过滤了差不多30%的基因
> dim(e);dim(filt1)
[1] 58037 108
[1] 40436 108
也有人自己写出了过滤代码
来自:https://seqqc.wordpress.com/2020/02/17/removing-low-count-genes-for-rna-seq-downstream-analysis/ By default evaluates to TRUE if >=90% (N=0.9 ) of the samples have count-per-million (CPM) above k, where k is determined by the default value of min.count=10 and by the sample library sizes.
这个过滤借助了genefilter包,比edgeR的更狠
selectGenes <- function(counts, min.count=10, N=0.90){
lib.size <- colSums(counts)
MedianLibSize <- median(lib.size)
CPM.Cutoff <- min.count / MedianLibSize*1e6
CPM <- edgeR::cpm(counts,lib.size=lib.size)
min.samples <- round(N * ncol(counts))
f1 <- genefilter::kOverA(min.samples, CPM.Cutoff)
flist <- genefilter::filterfun(f1)
keep <- genefilter::genefilter(CPM, flist)
## the same as:
#keep <- apply(CPM, 1, function(x, n = min.samples){
# t = sum(x >= CPM.Cutoff) >= n
# t
#})
return(keep)
}
keep.exprs <- selectGenes(assay(x), min.count=10, N=0.90)
myFilt <- x[keep.exprs,]
> dim(myFilt)
[1] 30467 108
使用TCGAanalyze_Filtering进行过滤
这个quantile参数留下的基因多一点,但是varFilter参数过滤的也太可怕了!
datNorm <- recount::scale_counts(x, by = "auc", round = FALSE)
filt2 <- TCGAanalyze_Filtering(assay(datNorm), method = "quantile")
> dim(filt2)
[1] 43527 108
datNorm <- recount::scale_counts(x, by = "auc", round = FALSE)
filt3 <- TCGAanalyze_Filtering(assay(datNorm), method = "varFilter")
> dim(filt3)
[1] 14509 108
关于method参数的解释:
- method == “quantile”: filters out those genes with mean across all samples, smaller than the threshold. The threshold is defined as the quantile of the rowMeans
qnt.cut = 0.25(by default 25% quantile) across all samples. - method == “varFilter”: This method filters out those genes with IQR across all samples, smaller than the threshold. The threshold is defined as the quantile of the rowIQRs
var.cutoff = 0.75(by default 75% quantile) across all samples. Here, the function IQR is used by default, but any other function can be used by changing the parametervar.func = IQR.
使用genefilter进行过滤
有没有发现,上面👆那个可怕的过滤varFilter,其实就是利用的这里genefilter 的一个函数
filt4 <- genefilter::varFilter(assay(datNorm), var.func = IQR,
var.cutoff = 0.75, filterByQuantile = TRUE)
> dim(filt4)
[1] 14509 108
当然可以调宽松一些:
filt5 <- genefilter::varFilter(assay(datNorm), var.func = IQR,
var.cutoff = 0.25, filterByQuantile = TRUE)
> dim(filt5)
[1] 43463 108
问题来了,这么多过滤方法,哪个更好?
上面展示了
filterByExpr,TCGAanalyze_Filtering,genefilter以及自定义的代码
其实这是一个伪问题:既然有不同方法,那就有不同参数,既然是参数,那就皆可调!
问题的关键不是选择什么方法,而是我们选择的基因数量。
如果你得到的基因数量很多,有用的也很多,那么多过滤一点也是无所谓;
但如果你本来的基因数量就比较可怜,还硬是要跟风去严格过滤,到头来后悔还是自己。