042-WGCNA
刘小泽写于18.10.10 很早就见过它,但只是不明觉厉,直到昨天才认真了解
是这样的:昨天看了一篇文献,讲冠心病的基因共表达分析,简而言之就是把得到的几千个变化比较大的基因和表型联系起来,将他们放入几十个模块中,最后通过比较模块、表型来确定要研究的那些基因,进而可以进行功能注释之类的下游分析。对于样本量大的研究(比如芯片、几十个样本的转录组、GWAS)来说,这么做是最快找到有价值基因的方法。
文章DOI:10.1186/s12872-016-0217-3
搜集资料
当然,所有pdf资料以及实战的教程数据都可以在公众号回复“WGCNA”得到
这是一个全新的领域,但是对R不陌生,上手会省时一些
搜索WGCNA tutorial 第一个就是啦。最新的教程还是16年更新的,看来作者对自己的作品比较满意啊,很少更新了现在
踏入新领域,首先就是要了解背景知识
WGCNA:全称 Weighted correlation network analysis,中文名是加权基因共表达网络分析
原来做几个样本的转录组,最后只要得到差异基因,再富集分析一下,就是一个经典的套路;但是样本数量如果很多呢?背景基因就会很多,再用传统的途径获得差异基因,往往会漏掉许多。WGCNA利用成千上万个变化显著的基因或全部基因的信息来筛选感兴趣的基因集,并且联系了表型的显著性关联分析。
这样做有两个好处:一个是损失的基因少;另一个是把众多的基因汇集到几个基因集,和表型进行关联,就不用做多重假设检验
Wighted加权/权重 衡量重要程度,也就是不仅要看基因是否相关,还要看相关性大小
Network 网络 简单网络:点和线组成,大部分点之间都有线连接 无尺度网络:大部分节点很少连接,少部分点占据大量连接。基因的调控关系就是无尺度网络
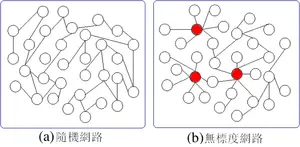
Correlation相关 依据相关性得到模块module ,指的是高度相关的基因,这些基因在不同组织或者一个生理过程中有相似的表达变化。每个模块中总会有佼佼者,也就是“第一成分”,就是说这个基因和许多基因都有联系,可以代表整体模块。相当于北上广这样的核心地位
走进它的世界
第一步 数据输入与初步处理
一般WGCNA需要15个样本以上的数据,样本越多结果越稳定
需要两样东西:表达矩阵datExpr、表型矩阵datTraits
表达矩阵: 芯片数据的归一化矩阵; 转录组的表达矩阵(一般以RPKM为单位)【一般都是基因在行,样品在列,后期还需要转置的】
表型矩阵:用于关联分析,必须是数值型矩阵 本身就是数值型:如长度、重量等,直接使用; 本身不是数值型,如患病与否这样的分类变量,需要用0-1表示(0表示没有该属性:患病,1表示有该属性),构建0-1矩阵后再分析
# 举个例子 ID Sick Height Weight sam1 1 1 2 sam2 1 2 7 sam3 0 10 22 sam4 0 NA 31 sam5 0 14 19
wkdir <- "."
setwd(wkdir)
library(WGCNA)
options(stringsAsFactors = FALSE)
femData = read.csv("LiverFemale3600.csv")
#dim(femData)
names(femData) #看下列名
进行转置:设置初步的表达矩阵:样本为行,观测为列
datExpr0 = as.data.frame(t(femData[, -c(1:8)]))
names(datExpr0) = femData$substanceBXH
rownames(datExpr0) = names(femData)[-c(1:8)]
检查基因和样本是否有太多的缺失值【太多就去掉】
gsg = goodSamplesGenes(datExpr0, verbose = 3)
gsg$allOK # 返回TRUE则继续
# (可选)如果存在太多的缺失值
if (!gsg$allOK)
{
# 把含有缺失值的基因或样本打印出来
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")));
# 去掉那些缺失值
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
对样本进行聚类,检测异常值
if(T){
sampleTree = hclust(dist(datExpr0), method = "average")
pdf(file = "sampleClustering.pdf", width = 12, height = 9)
par(cex = 0.6)
par(mar = c(0,4,2,0))
plot(sampleTree, main = "Sample clustering to detect outliers", sub="", xlab="", cex.lab = 1.5,
cex.axis = 1.5, cex.main = 2)
# 结果可以看到,F2_221是一个异常值
abline(h = 15, col = "red") #先画一条辅助线
dev.off()
}

去除异常值,得到过滤后的表达矩阵
# 把高于15的切除
clust = cutreeStatic(sampleTree, cutHeight = 15, minSize = 10)
table(clust) # 0代表切除的,1代表保留的
keepSamples = (clust==1)
datExpr = datExpr0[keepSamples, ]
开始设置表型矩阵=》加载表型信息
再次注意:所有的表型信息都要用数值来表示在各个样本中的大小 (比如:体重、长度、肥胖度等都要量化)
traitData = read.csv("ClinicalTraits.csv")
#dim(traitData)
names(traitData)
# 去掉不需要的信息,得到全部样本的表型矩阵
allTraits = traitData[, -c(31, 16)] #去掉'note'、'comments'
allTraits = allTraits[, c(2, 11:36) ] # 去掉中间的日期之类的
dim(allTraits)
names(allTraits)
得到部分表型矩阵=》表达矩阵有哪些样本,表型矩阵就有哪些样本
femaleSamples = rownames(datExpr)
traitRows = match(femaleSamples, allTraits$Mice)
# 把原来的第一列Mice变成行名
datTraits = allTraits[traitRows, -1]
rownames(datTraits) = allTraits[traitRows, 1]
collectGarbage() #释放内存空间
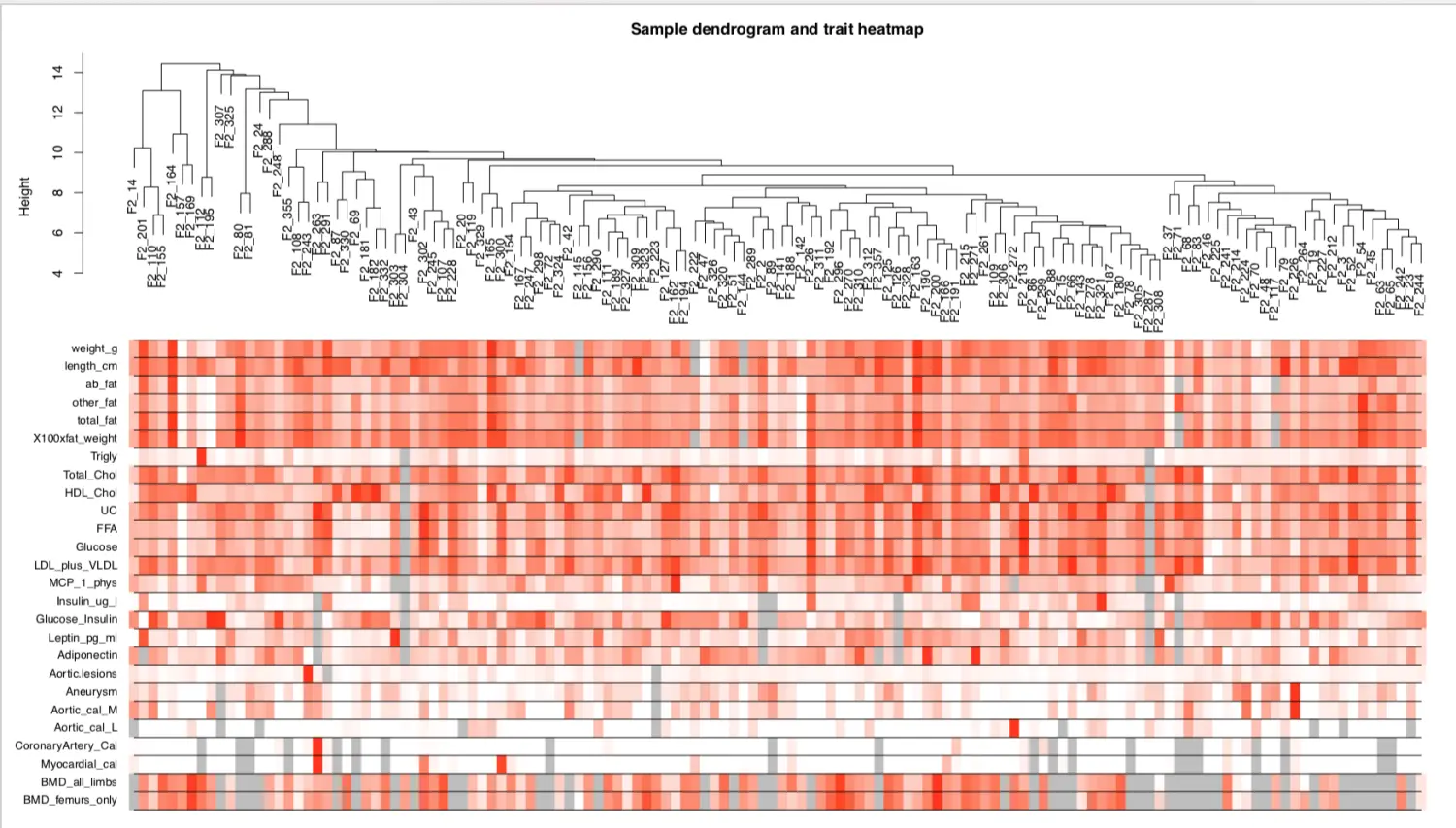
到这里为止,得到了所有样本的表达矩阵和表型矩阵,在进一步构建网络、检测模块之前,先看下表型和样本的相关性
针对去除异常值的表达矩阵进行聚类分析
sampleTree2 = hclust(dist(datExpr), method = "average")
# 用颜色表示表型在各个样本的表现: 白色表示低,红色为高,灰色为缺失
traitColors = numbers2colors(datTraits, signed = FALSE)
# 把样本聚类和表型热图绘制在一起
if(T){
pdf(file = "Sample dendrogram and trait heatmap.pdf", width = 18, height = 10)
plotDendroAndColors(sampleTree2, traitColors,
groupLabels = names(datTraits),
main = "Sample dendrogram and trait heatmap")
dev.off()
}
save(datExpr, datTraits, file = "FemaleLiver-01-dataInput.RData")
第二步 网络分析
构建网络、检测协同表达的基因模块**(非常重要的一步!)**
实现构建网络有3种方法:(3选1即可) 1.One-step; 2.Step-by-step 支持自定义一些参数; 3.Block-wise network construction 针对大型数据
加载之前保存的RData
lnames = load(file = "FemaleLiver-01-dataInput.RData")
这里采用一步分析法
筛选软阈值(soft thresholding power)
原则是使构建的网络更符合无标度网络特征
#设置一系列软阈值(默认1到30)
powers = c(c(1:10), seq(from = 12, to=20, by=2))
#帮助用户选择合适的软阈值,进行拓扑网络分析
#需要输入表达矩阵、设置的阈值范围、运行显示的信息程度(verbose=0不显示任何信息)
sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
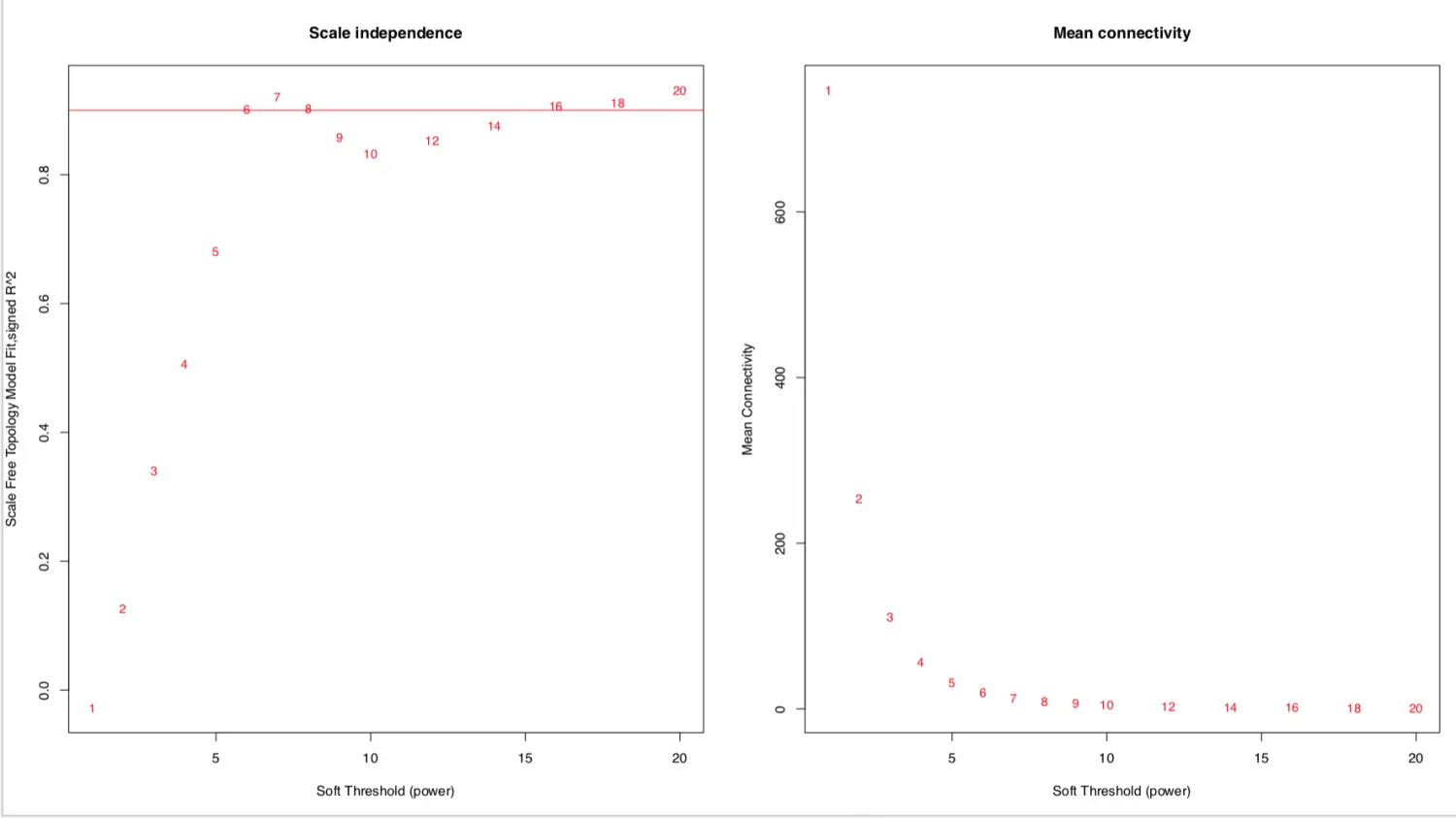
sft结果有两列,其中powerEstimate返回最佳的软阈值beta值。这里的结果是6,表示从6达到高阈值以后,图形曲线开始变平,就是说:到达阈值6时网络拓扑结构连通的就差不多了,够本了!具体见下面做出的图(左一)来理解。实际使用并不需要知道具体值
画出结果 =》横轴是Soft threshold (power) **(左图)**纵轴是无标度网络的评估参数(相关系数的平方),数值越高,网络越符合无标度特征 (non-scale) **(右图)**纵轴是基因模块中所有基因邻接函数的均值
if(T){
pdf(file = "Soft threshold.pdf", width = 18, height = 10)
par(mfrow = c(1,2))
cex1 = 0.9
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",
ylab="Scale Free Topology Model Fit,signed R^2",type="n",
main = paste("Scale independence"))
#注意这里的-sign(sft$fitIndices[,3])中sign函数,它把正数、负数分别转为1、-1
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red")
# 设置筛选标准h=r^2^=0.9。这里的0.9是个大概的数,就是看左图软阈值6大概对应的位置
abline(h=0.90,col="red")
#看一下Soft threshold与平均连通性
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",
main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")
dev.off()
}
一步构建网络[关键一步!]
几千个基因组归类成了几十个模块
计算基因间的邻接性,根据邻接性计算基因间的相似性,然后算出基因间的相异性系数,并因此得到基因间的系统聚类树 按照混合动态剪切树的标准,设置每个基因模块最少的基因数目为30 确定基因模块后,再次分析,依次计算每个模块的特征向量值 对模块进行聚类分析,将距离较近的模块合并为新的模块 power就是上面计算得到的软阈值
net = blockwiseModules(datExpr, power = 6,
TOMType = "unsigned", minModuleSize = 30,
reassignThreshold = 0, mergeCutHeight = 0.25,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "femaleMouseTOM",
verbose = 3)
# 显示模块数量以及各自包含的基因数目
# 0表示未分入任何模块的基因
# 1是最大的模块,往后依次降序排列,分别对应各自模块的基因
table(net$colors)
模块可视化=》聚类树
将每个模块对应的基因数转换成颜色单位, 灰色默认是无法归类于任何模块的那些基因
mergedColors = labels2colors(net$colors)
先画聚类,后画颜色
if(T){
pdf(file = "DendroAndColors.pdf", width = 18, height = 10)
plotDendroAndColors(net$dendrograms[[1]], mergedColors[net$blockGenes[[1]]],
"Module colors",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
dev.off()
}
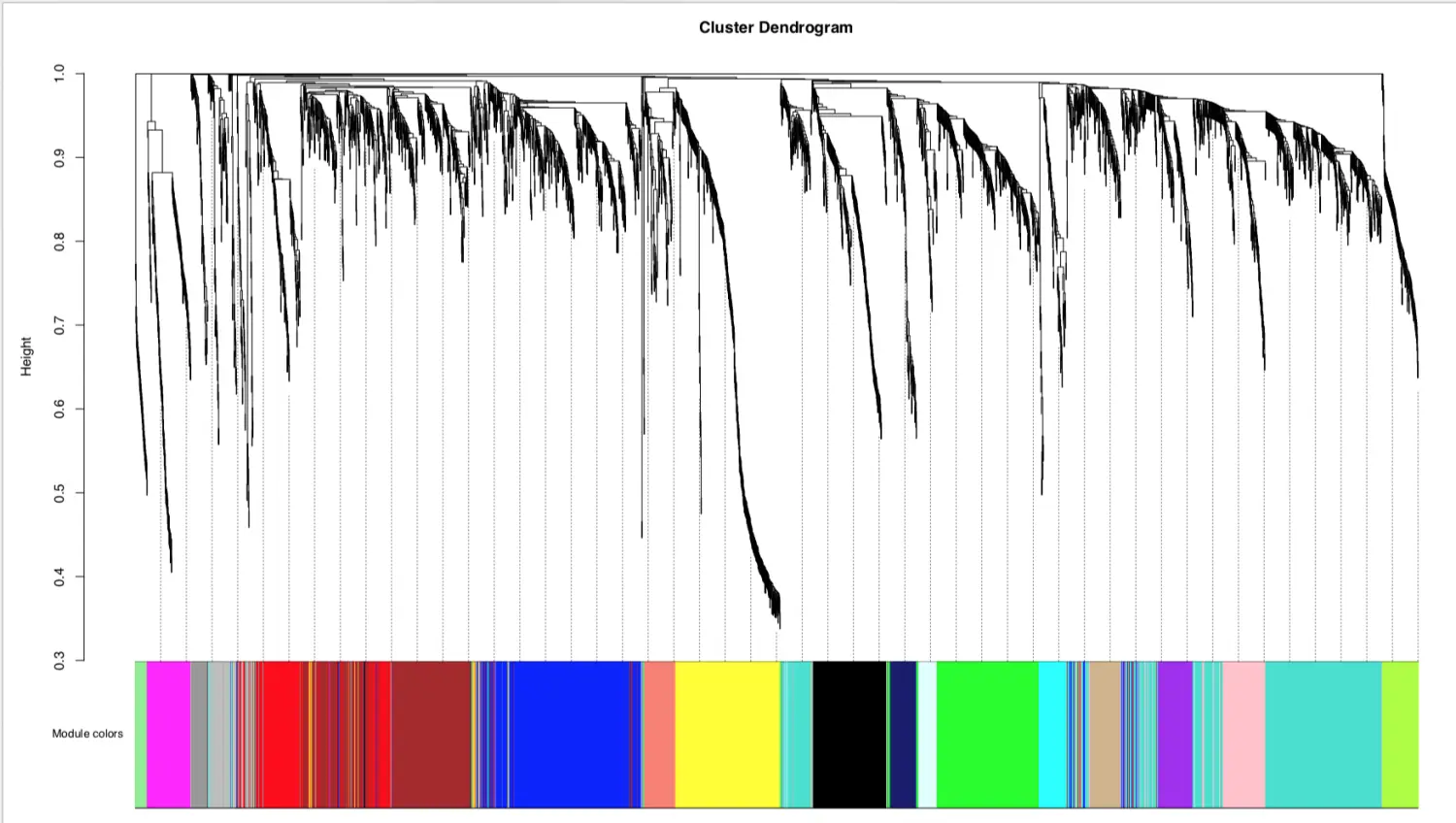
保存数据
moduleLabels = net$colors
moduleColors = labels2colors(net$colors)
MEs = net$MEs
geneTree = net$dendrograms[[1]]
save(MEs, moduleLabels, moduleColors, geneTree,
file = "FemaleLiver-02-networkConstruction-auto.RData")
第三步 模块联系表型信息
看看哪些表型的哪些模块是自己感兴趣的(一般选热图颜色深的)
这样初步表明该模块中基因可能是有研究价值的; 下一步,进行一个验证,看看这个模块中基因与表型、模块的相关性,看看与模块相关的基因是不是也与表型相关
rm(list = ls())
load(file = "FemaleLiver-01-dataInput.RData")
load(file = "FemaleLiver-02-networkConstruction-auto.RData")
模块关联表型
# 得到基因、样本数量
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
# 用color labels重新计算MEs(Module Eigengenes:模块的第一主成分)
MEs0 = moduleEigengenes(datExpr, moduleColors)$eigengenes
MEs = orderMEs(MEs0)
moduleTraitCor = cor(MEs, datTraits, use = "p") #(这是重点)计算ME和表型相关性
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples)
对moduleTraitCor画热图,看结果挑选自己感兴趣的模块进行下游分析
if(T){
pdf(file = "labeledHeatmap.pdf", width = 18, height = 10)
# 设置热图上的文字(两行数字:第一行是模块与各种表型的相关系数;
# 第二行是p值)
# signif 取有效数字
textMatrix = paste(signif(moduleTraitCor, 2), "\n(",
signif(moduleTraitPvalue, 1), ")", sep = "")
dim(textMatrix) = dim(moduleTraitCor)
par(mar = c(6, 8.5, 3, 3))
# 然后对moduleTraitCor画热图
labeledHeatmap(Matrix = moduleTraitCor,
xLabels = names(datTraits),
yLabels = names(MEs),
ySymbols = names(MEs),
colorLabels = FALSE,
colors = greenWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
main = paste("Module-trait relationships"))
dev.off()
}
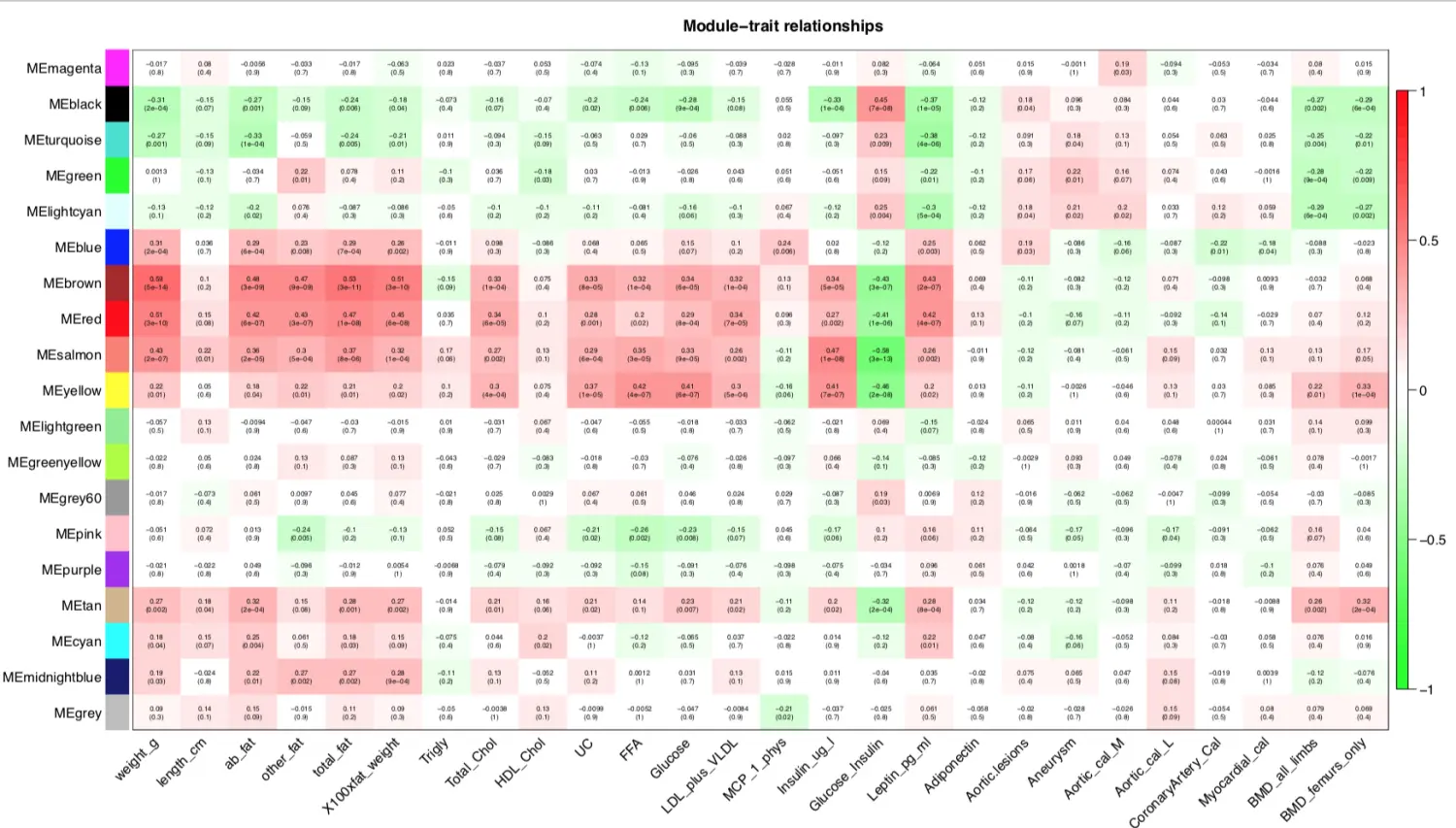
每种表型都有和它非常相关的模块,因此某个模块可以作为某个表型的代表(signature),对于非常相关的模块,比如weight表型和brown模块,颜色最深。**那么里面的基因是什么?**于是进行感兴趣表型中核心模块的基因分析
计算基因与模块的相关性矩阵(MM: Module Membership)
# 把各个module的名字提取出来(从第三个字符开始),用于一会重命名
modNames = substring(names(MEs), 3)
# 得到矩阵
geneModuleMembership = as.data.frame(cor(datExpr, MEs, use = "p"))
# 矩阵t检验
MMPvalue = as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nSamples))
# 修改列名
names(geneModuleMembership) = paste("MM", modNames, sep="")
names(MMPvalue) = paste("p.MM", modNames, sep="")
计算基因与表型的相关性矩阵(GS: Gene Significance)
# 先将感兴趣表型weight提取出来,用于计算矩阵
weight = as.data.frame(datTraits$weight_g)
names(weight) = "weight"
# 得到矩阵
geneTraitSignificance = as.data.frame(cor(datExpr, weight, use = "p"))
# 矩阵t检验
GSPvalue = as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance), nSamples))
# 修改列名
names(geneTraitSignificance) = paste("GS.", names(weight), sep="")
names(GSPvalue) = paste("p.GS.", names(weight), sep="")
合并两个相关性矩阵,找到和模块、表型都高度相关的基因
if(T){
module = "brown"
pdf(file = paste0(module,"-MM-GS-scatterplot.pdf"), width = 10, height = 10)
column = match(module, modNames) #找到目标模块所在列
moduleGenes = moduleColors==module #找到模块基因所在行
par(mfrow = c(1,1))
verboseScatterplot(abs(geneModuleMembership[moduleGenes, column]),
abs(geneTraitSignificance[moduleGenes, 1]),
xlab = paste("Module Membership in", module, "module"),
ylab = "Gene significance for body weight",
main = paste("Module membership vs. gene significance\n"),
cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module)
dev.off()
}

图中看到,MM和GS高度正相关,因此说明和表型高度相关的基因,在与表型相关模块中也是核心元素。
另外,也可以探索weight表型中其他的模块,比如第一个颜色很浅的magenta模块,它的MM和GS整体负相关
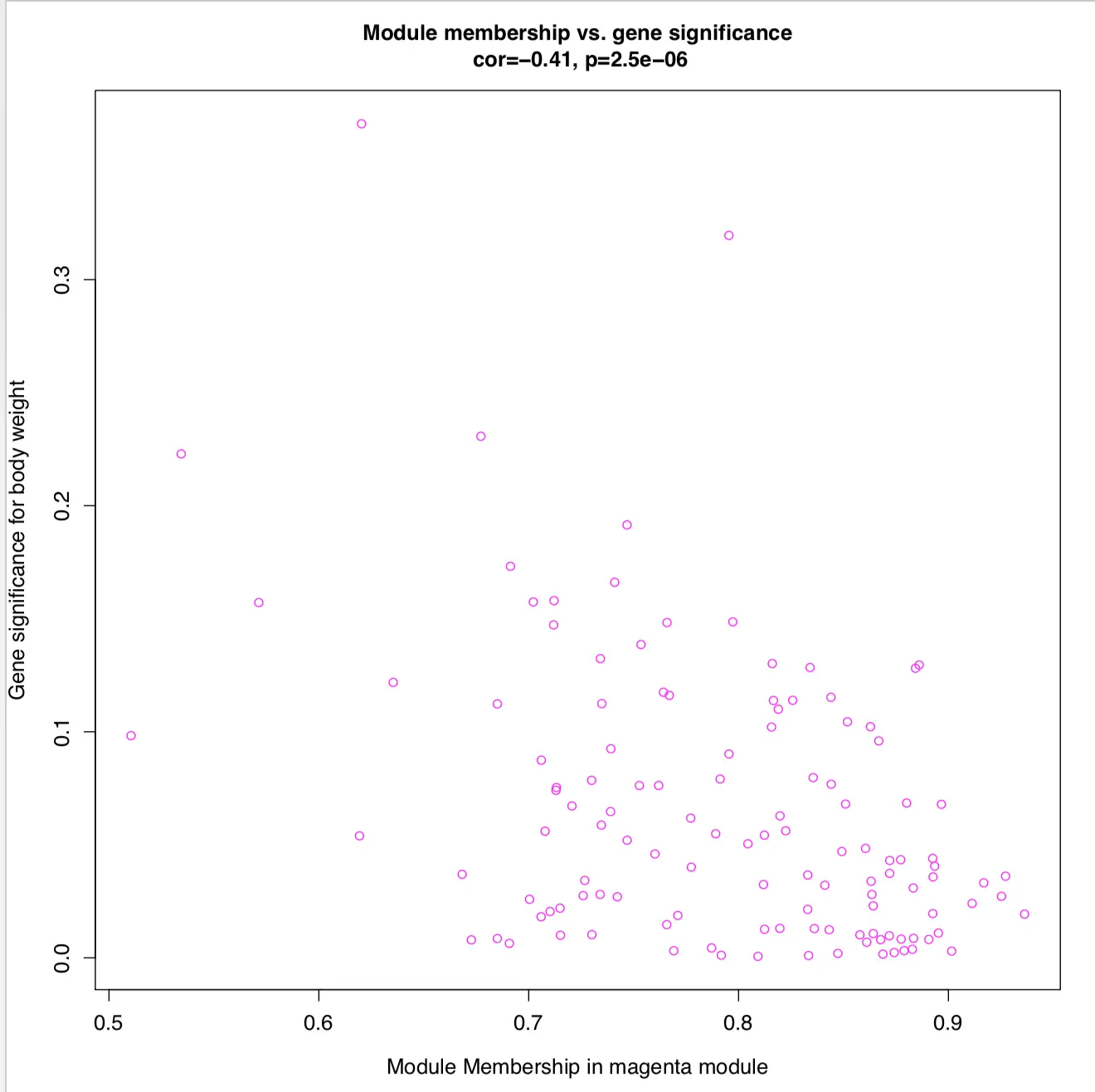
第四步 网络可视化
【针对基因】热图的方式展示加权网络,每行每列代表一个基因
还能表示邻近关系和拓扑重叠,浅颜色表示关系弱,深颜色表示关系强
基因的聚类分析和模块颜色画在顶部和左侧
rm(list = ls())
load(file = "FemaleLiver-01-dataInput.RData")
load(file = "FemaleLiver-02-networkConstruction-auto.RData")
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
选择400个基因画图
if(T){
nSelect = 400
set.seed(10)
# 计算拓扑重叠(TOM: Topological Overlap Matrix)
# 这个过程先计算了邻接矩阵,后把邻接矩阵转换为拓扑重叠矩阵,
# 降低了噪音和假相关,获得距离矩阵dissTOM
dissTOM = 1-TOMsimilarityFromExpr(datExpr, power = 6)
select = sample(nGenes, size = nSelect)
selectTOM = dissTOM[select, select]
# 再计算基因之间的距离树(对于基因的子集,需要重新聚类)
selectTree = hclust(as.dist(selectTOM), method = "average")
selectColors = moduleColors[select]
pdf(file = paste0("Sub400-netheatmap.pdf"), width = 10, height = 10)
plotDiss = selectTOM^7
diag(plotDiss) = NA #将对角线设成NA,在图形中显示为白色的点,更清晰显示趋势
TOMplot(plotDiss, selectTree, selectColors, main = "Network heatmap plot, selected genes")
dev.off()
}
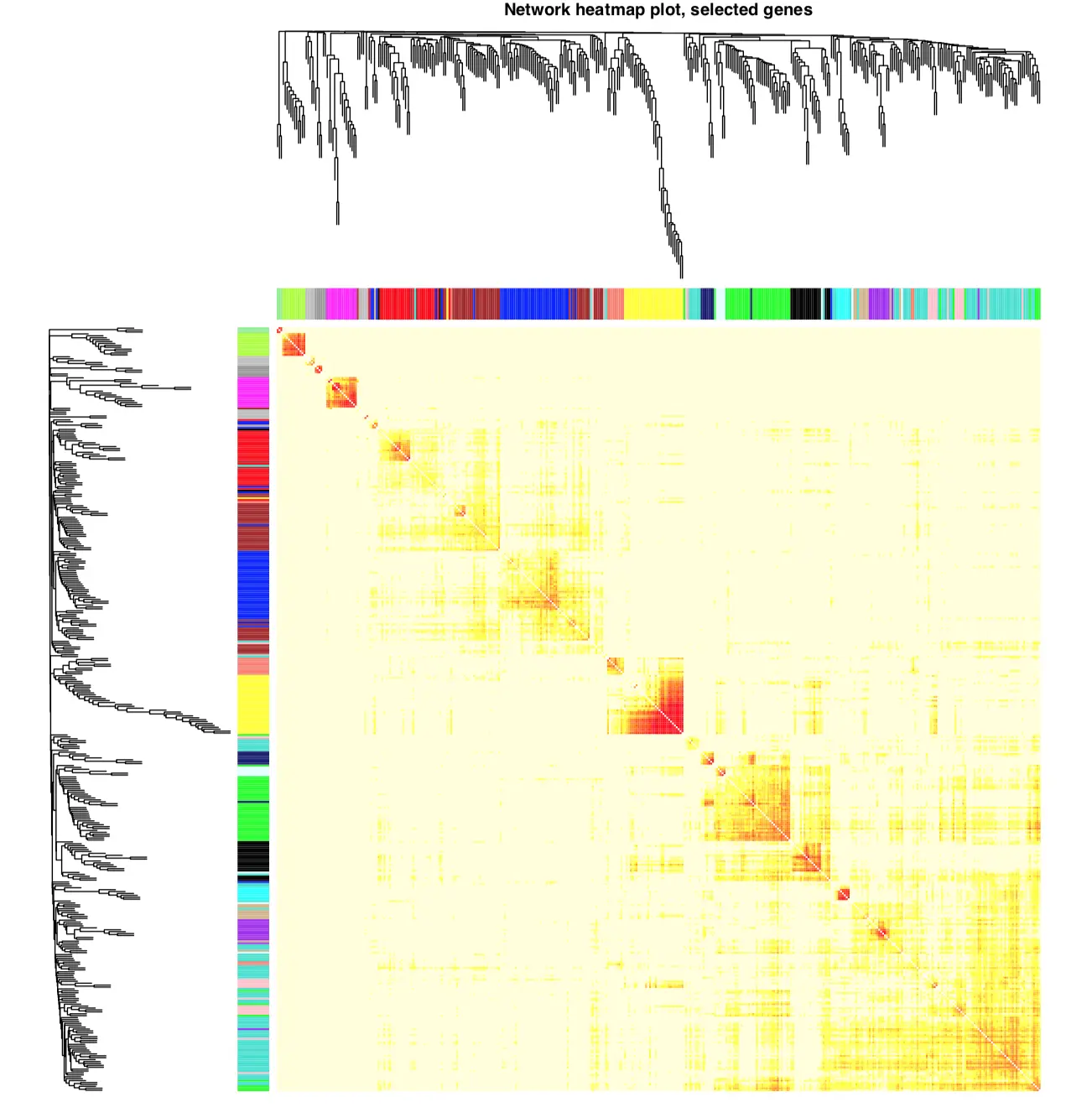
选择全部基因画图(耗时较久,生成的文件很大)
结果可以看到各个区块的颜色差异,沿着对角线的深色区块就是模块Module
if(T){
pdf(file = "All-netheatmap.pdf", width = 10, height = 10)
plotTOM = dissTOM^7
diag(plotTOM) = NA
TOMplot(plotTOM, geneTree, moduleColors, main = "Network heatmap plot, all genes")
dev.off()
}
【针对模块与表型】展示模块与表型的相关性
重新计算模块的基因样本相关矩阵(Eigengenes就是基因和样本的相关矩阵)
MEs = moduleEigengenes(datExpr, moduleColors)$eigengenes
# 将weight表型信息从trait中提取出来
weight = as.data.frame(datTraits$weight_g)
names(weight) = "weight"
# 把weight表型添加到之前计算的ME矩阵中,并排序
MET = orderMEs(cbind(MEs, weight))
# 画图=》meta-modules(模块的聚类图加上模块与表型的热图)
# marDendro/marHeatmap 设置下、左、上、右的边距
if(T){
pdf(file = "Eigengene-dengro-heatmap.pdf", width = 10, height = 15)
par(cex = 0.9)
plotEigengeneNetworks(MET, "", marDendro = c(0,4,1,2), marHeatmap = c(4,4,1,2), cex.lab = 0.8, xLabelsAngle
= 90)
dev.off()
}
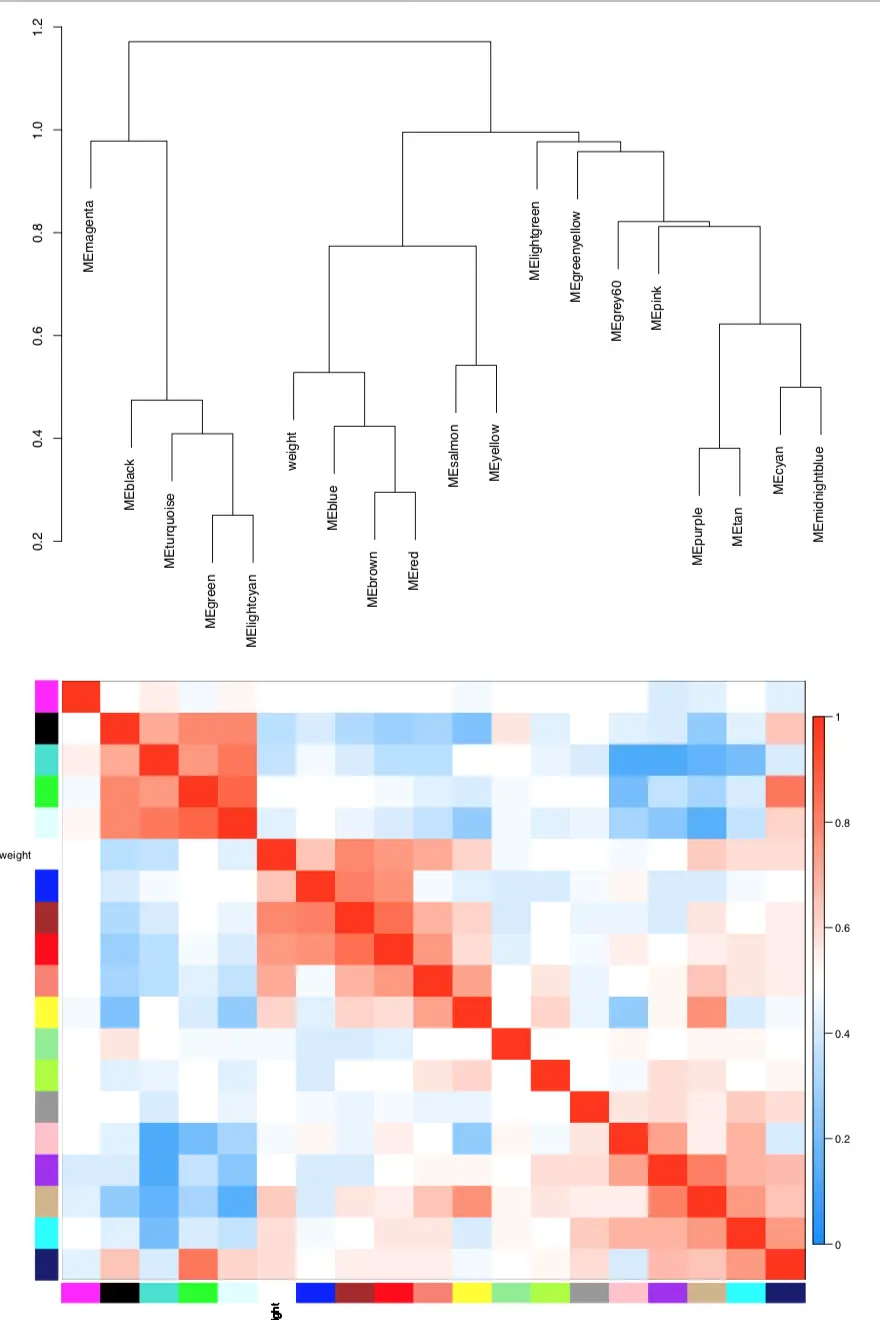
图中可以看到,red、blue、brown模块是高度相关的,并且它们的相关性比各自和weight表型的相关性还大;salmon和weight的相关性也比较强,但是加入red、blue、brown的meta-modules阵营还不够格
当然分开画也是可以的
if(T){
pdf(file = "Eigengene-dendrogram.pdf", width = 10, height = 10)
par(cex = 1.0)
plotEigengeneNetworks(MET, "Eigengene dendrogram", marDendro = c(0,4,2,0),
plotHeatmaps = FALSE)
dev.off()
pdf(file = "Eigengene-heatmap.pdf", width = 10, height = 10)
par(cex = 1.0)
plotEigengeneNetworks(MET, "Eigengene adjacency heatmap", marHeatmap = c(4,5,2,2),
plotDendrograms = FALSE, xLabelsAngle = 90)
dev.off()
}
第五步 导出网络
准备过程
# 重新计算拓扑重叠矩阵
TOM = TOMsimilarityFromExpr(datExpr, power = 6)
# 选择导出模块
module = "brown"
# 选择模块中基因/探针
probes = names(datExpr)
inModule = (moduleColors==module)
modProbes = probes[inModule]
# 选择相关模块的拓扑重叠矩阵
modTOM = TOM[inModule, inModule]
dimnames(modTOM) = list(modProbes, modProbes)
导出到VisANT
vis = exportNetworkToVisANT(modTOM,
file = paste("VisANTInput-", module, ".txt", sep=""),
weighted = TRUE,
threshold = 0)
感觉基因太多,可以截取部分(比如前30)
nTop = 30
IMConn = softConnectivity(datExpr[, modProbes])
top = (rank(-IMConn) <= nTop)
submodTOM <- modTOM[top, top] #然后用submodTOM代替前面的modTOM导出
导出到Cytoscape
modules = c("brown", "red")
cyt = exportNetworkToCytoscape(modTOM,
edgeFile = paste("CytoscapeInput-edges-", paste(modules, collapse="-"), ".txt", sep=""),
nodeFile = paste("CytoscapeInput-nodes-", paste(modules, collapse="-"), ".txt", sep=""),
weighted = TRUE,
threshold = 0.02,
nodeNames = modProbes,
nodeAttr = moduleColors[inModule])
# 同理,感觉基因太多可以用submodTOM代替modTOM
写在最后
这只是一个基础的流程,为了熟悉熟悉流程。只是针对一个大样本;如果有多个来源不同的大样本进行分析,还需要参考官网的第二部分教程
得到了模块中的基因后,就可以进行GO、KEGG的富集分析,可以挖掘模块中关键基因,然后预测基因功能