045-简单理解RNA-seq
刘小泽写于2018.10.17 今天就当一个小故事看吧,看了statQuest,感觉讲的很棒,于是分享给大家
https://www.youtube.com/watch?v=tlf6wYJrwKY&list=PLblh5JKOoLUIcdlgu78MnlATeyx4cEVeR&index=6
背景 1
假设有一群正常的神经细胞(蓝色)和一群变异的神经细胞(红色)

那么,为什么它们会出现差别呢?是什么遗传机制导致了这个事情呢?
因此,我们需要看一看它们的基因表达差异
背景 2
我们知道,每个细胞都由一堆染色体组成,每个染色体由一堆基因组成,当然并不是所有的基因都是活跃的,只有一部分基因是可以表达,而表达的中间过程就要经历mRNA转录本,通过高通量测序,我们就能得知:哪些基因是活跃可以表达的,并且产生了多少转录本(也就是衡量基因表达量的指标)
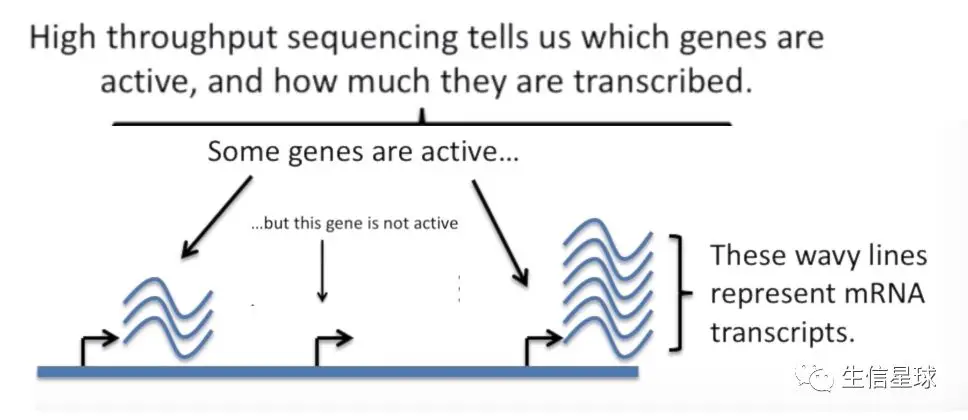
背景 3
将正常的细胞测一遍,再将变异的细胞测一遍,得到它们的表达量,我们后来就是比较它们的表达量差异
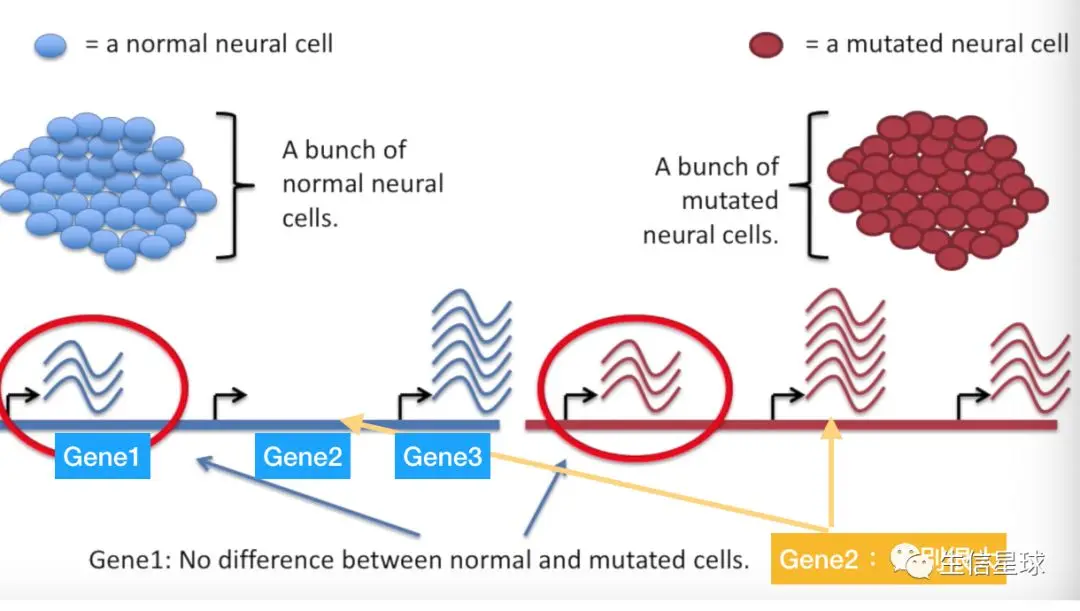
可以看出,基因1在两组样本中差异不大或者没有差异;基因2在正常组中基本不表达,而在变异组中表达量很高,二者差别甚大;基因3有差别但比较小
RNA-seq主要的3步
Step1 构建测序文库
分离RNA=》将RNA打断成小片段=〉将小RNA片段反转录成DNA=》加接头
接头两个作用:测序仪识别;允许一台测序仪同时运行多个样本,提高性价比
但是需要注意:加接头的过程是随机的,并不是所有的接头都被加上,有些反转录的DNA片段没有加上接头
=》PCR扩增(只有加上接头的测序片段才能被扩增)=〉质量检查QC(看下文库的浓度和片段长度)
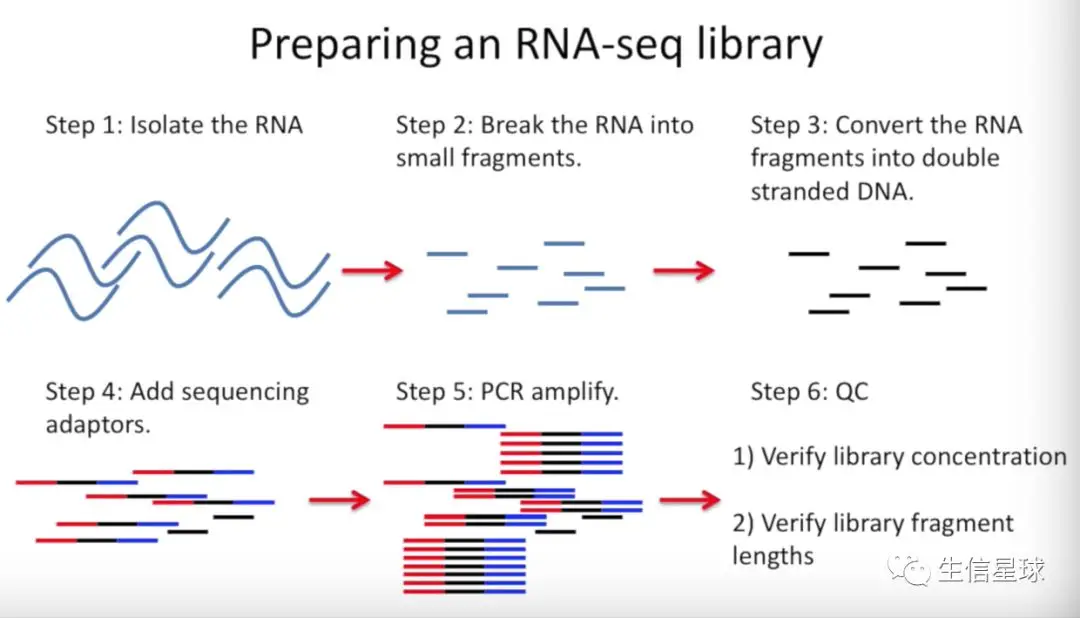
对文库进行测序
一块测序板上(想象下载玻片,其实人家真名是Flowcell)能包含多于400,000,000个片段,垂直于测序板排列。
测序仪有四种颜色的荧光探针A、T、C、G,与测序片段上碱基互补,结合上就“放烟花”表示庆祝🎉(就是闪一下自己带的荧光,比如A带红光,G带蓝光,C绿光,T橙光)。当然,这一切都逃不过测序仪自带的高精度照相机的法眼【测序仪为什么贵?就是在于它的高精度照相机,想想要分辨这么微小的亮光,密密麻麻,密集恐机症都犯了🤢】许许多多的测序片段中同一排的碱基测完了,就把原来荧光的那个碱基冲掉了,再放下一个荧光碱基进来结合、放光
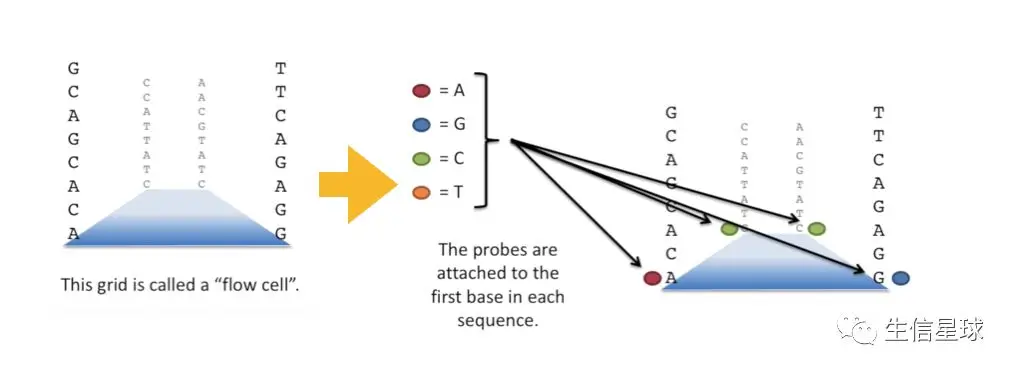
测序就是这样,结果就得到了raw data,就是fastq数据
Step 2 原始数据处理
质控=》过滤garbage reads=〉比对到参考基因组=》再数一下每个基因比对上多少reads
garbage reads:
有些时候接头并没有加到测序片段,而是他们直接结合,也能进行测序,但测得结果是没用的
比对到参考基因组
先将大的基因组序列打断成许多小片段,然后为了方便接下来寻找这些片段,需要对他们进行构建索引index(目的就是标注每个小片段的位置)
再将测序的reads和基因组一样,也是打断成小片段,然后把它的小片段比对到基因组的小片段上,比对上的会给出位置信息

统计reads数得到表达矩阵
就想这样:第一列是基因名(人类基因组有大概2w基因,因此大概有2w行)
其他列是每个测序样本比对上的数量(6-成百上千不等),这里的6的考虑的是处理对照各3个重复,即Bulk-seq;大样本量的RNA-seq比如Single-cell,每个细胞都是一个样本,因此成百上千
每一行都是原始的统计值,每个基因在每个样本中被抓到多少次

标准化表达矩阵
进行标准化的原因是:某些样本可能本身测序质量就差,但并不代表人家没东西;或者有的样本测序的时候加的浓度比较高,因此统计时占优势,但并不公平!
因此需要让大家在同一起跑线

Step 3 可视化
比如PCA分析,看看样本之间能否区分开,另外可以排除明显不对的样本,比如这里的wt2

然后看差异表达基因(就是正常与变异样本的差异)
红色是差异的,黑色是共同的

如果发现了感兴趣的差异基因,怎么办?
- 这个基因是你研究的,接下来通过实验验证
- 对这个基因不熟悉,只是感兴趣,就可以做GO、KEGG注释,看看它在正常还是变异样本中有富集