046-标准化进行时
刘小泽写于18.10.19 今天看一看转录组的定量相关术语
首先看概念
我们做转录组很大程度上是为了看基因表达量差异的,那么什么是表达量? 它肯定是用来看两组之间不同的一个标准,但这个标准怎么定义呢?
一个细胞中或者一定含量的RNA中,含有转录本的数目就是表达量
进行差异分析之前,我们必须要先知道每个基因各自在不同样本中有多少,也就是raw counts。当然有的软件可以直接用raw counts进行比较,(例如DeSeq2就要求表达量矩阵是整数组成的、未经下游标准化的)。但是还有一些是需要标准化以后才能用的比如edgeR,那么什么是标准化?
我们赛跑都要讲求公平吧,比较基因表达量也是如此。表达量受到测序深度和基因长度的影响
同一个基因在不同样本中,样本测序深度越高,也就相当于抽样的次数越多,当然抽到落在该基因的reads数就越多;
同一个样本中的不同基因,基因越长,被打断后得到的片段就越多,会有越多的reads会落在它上面;
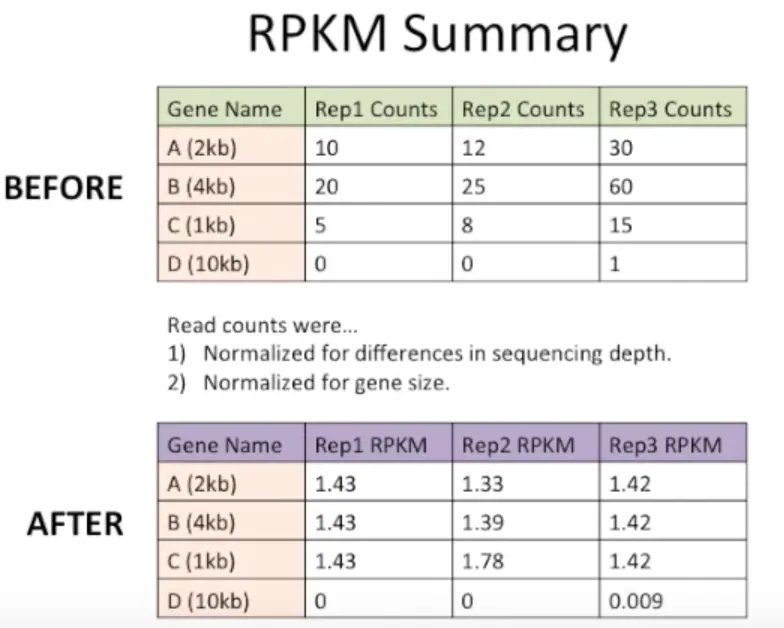
为了让不同样本的基因或者不同基因在同一样本中进行比较,需要把它们放在统一的起跑线上 于是要把原始的表达量统计值转换成RPKM/FPKM/TPM等,来消除测序深度和基因长度的影响
过去一般使用RPKM(Reads Per Kilobase Million)或者FPKM(Fragments Per Kilobase Million)
好吧,不得不说这个命名让人很迷惑 深入了解一下:首先,M和K都是放在分母上的, 其次,上面的M也就是Million,用来排除测序的深度的影响;K也就是Kilobase排除是基因的长度的影响 最后,公式的意思也就清楚啦:就是用reads原始统计值处以总reads数和基因长度
TPM(Transcripts per million)是更为推荐的
理解标准化
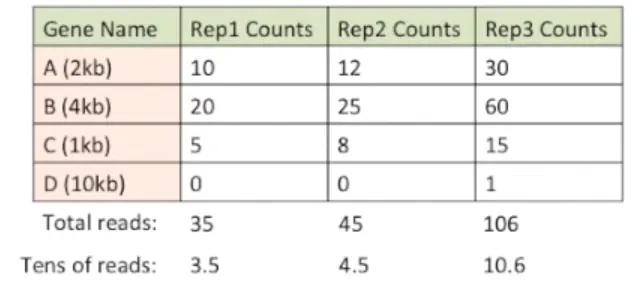
假如一开始得到的raw counts是这样的:一个样本3个重复,检测了4个基因
| 基因名 | 样本1.1 | 样本1.2 | 样本1.3 |
|---|---|---|---|
| A(2kb) | 10 | 12 | 30 |
| B(4kb) | 20 | 25 | 60 |
| C(1kb) | 5 | 8 | 15 |
| D(10kb) | 0 | 0 | 1 |
很明显,样本1.3中每个基因的值都明显高于1.1和1.2,另外B在所有样本中表达量都是A的2倍,当然,这些都是需要进行标准化的。看看标准化后是什么样子
- 利用RPKM
首先,将测序深度进行标准化:计算每列的总和然后除以1,000,000【当然这里由于是编的数据,就使用10进行整除】
然后,将基因长度标准化,就是将上一步数据再中心化(scale函数可以做到)
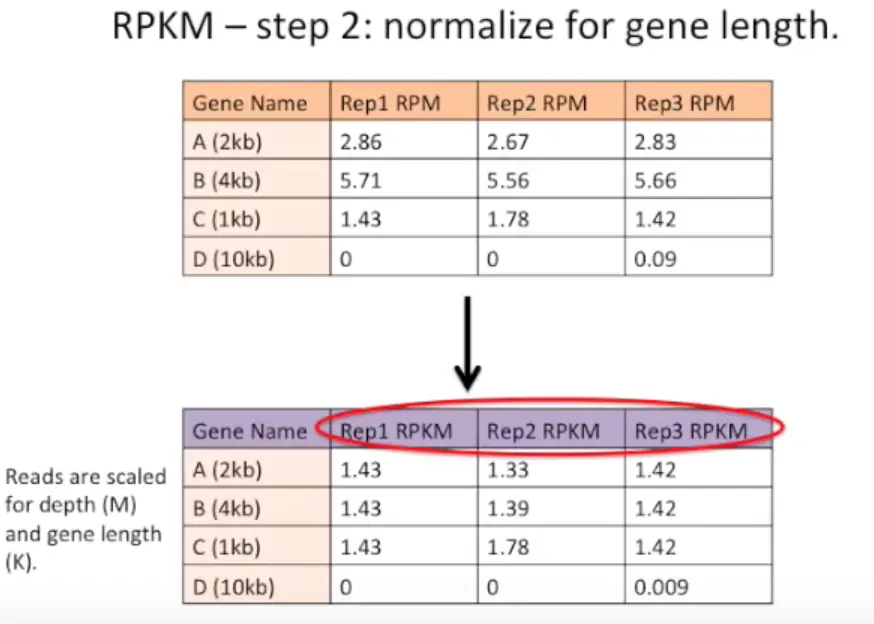
最后得到的就是这样啦
RPKM vs FPKM
只要知道:RPKM是单端数据使用,双端测序使用FPKM更合适
TPM
先对每个基因的reads数用基因长度校正,然后用校正后的reads数除以所有reads总和,因此每个样本中的TPM的总和都是一样的,而FPKM则不相等

结论
RPKM及TPM的都考虑了基因长度,但基因长度对RNA-Seq差异比较结果并无极大影响。单一样本中TPM与RPKM的结果基本上是一致的,同一物种不同的样本FPKM标准化会相对弱一些(也就是相对粗放一些),不同物种的就不能比较啦。 更推荐使用TPM进行标准化,但现实情况是:FPKM使用仍然比较广泛
刘小泽写于18.10.20
昨天写了关于基因表达量的一些术语,那么你是否会好奇,这些定量的值是怎么来的呢?我们又如何将原始的统计值raw counts转为标准化的FPKM或者TPM呢
表达量的意义
基因表达量事关重大,不同的表达量会产生不同的基因产物,也就是蛋白质啦。蛋白质又会决定细胞以及个体的形态与生物功能。因此转录组才会这么火热,因为我们都想探究某一种表型的内在原因,看看是哪些基因起主导作用,是促进还是抑制
来自哪里—检测表达量
我们对表达量最直观的就是检测,有的使用实验方法+图像,有的是用数据展示,有的方法适用于少部分基因检测(一般就是实验方法),有的用的大样本多基因检测
| 检测方法 | 原理 | 实际应用 |
|---|---|---|
| qPCR(实时荧光定量) | PCR扩增过程中增加荧光信号的实时监测,看看每个循环产物数量的变化 | 常规基因表达验证(低通量) |
| Northern blotting | 放射性标记探针杂交 | 半定量分析(用于少量基因的定性)(低通量) |
| FISH(荧光原位杂交) | 在组织水平上利用目的蛋白的抗体检测表达 | 某个基因在特定组织中表达(低通量) |
| SAGE(基因表达系列分析) | 分析大量的EST(表达序列标签)寻找不同丰度的标签序列 | 分析样本量较多(中通量) |
| Microarray(表达芯片或者微阵列) | 将寡核苷酸探针固定在芯片上,然后将待测样本mRNA加上荧光标记与芯片杂交,然后通过分析荧光信号来监测表达量 | 高通量范围 |
| RNA-seq | 直接就是高通量测序,将个体的RNA序列测出然后用比对的方法鉴定表达量 | 高通量范围 |
高通量的表达量来源之一:芯片
原来的芯片技术可谓是“一枝独秀”,一张芯片可能包含数十、数百甚至数十万的探针,利用芯片数据可以帮助发现有关键生物功能的基因,并对基因给出注释。并且对计算机要求不高,普通电脑(8G+内存)利用R的bioconductor就可以分析,因此在RNA-seq如此流行的今天,仍然有大量的芯片数据存放在NCBI的GEO数据库和EBI的ArrayExpress数据库等待解读,这些数据主要来自Affymetrix、Agilent、Illumina公司
芯片表达量的来源大体是这样的: 1.样本提取mRNA=》2.反转录成cDNA=〉3.荧光或同位素标记=》 4.液相环境中与芯片探针杂交=〉洗膜=》 5.扫描仪扫描荧光或同位素信号=〉原始数据获得=》 6.预处理(过滤背景噪音、数据筛选)=〉基因表达数据(每行是一个基因表达量,每列是样本所有基因)
然后再介绍下其中的关键几点
- 过滤背景“噪音”:这个噪音不是真的声音啦,而是图像中识别不清楚的。图像处理软件先对芯片划分单元,然后将整个芯片杂交点外的平均吸光度值作为背景,也就是过滤标准
- 筛选是怎么筛选的?
- 首先按点进行筛选,可以根据点的大小(size)、标记(flag)以及信号强度(intensity),筛选出来用缺失值NA替换原有值;
- 然后标准化或者归一化(normalization),这个就是为了分析不同的芯片结果或者同一芯片不同标记的两个样本数据。根据芯片通道不同,标准化方法也不同。 假如有三张芯片要比较,单通道只能选一张芯片作参照,其他芯片按参照进行中位数标准化(median)或管家基因标准化(housekeeping gene); 双通道需要对每张芯片都进行标准化,除了单通道的两种标准化方式,还可以用点样组内标准化、Lowess 标准化
- 去掉异常值(truncation),设置一个信号阈值,超过的要被截掉(主要用于双通道)
- 筛选基因:上面👆三步都是按质量过滤,这一步要按信息量过滤。先制定一个标准,除掉含有信息较少的基因(三种方式:用对数Log expression variance filter、用空缺百分比Missing percent filter、用最小变化倍数Minimum fold-change filter)
高通量的表达量来源之二:转录组
RNA-seq表达量来源 大体是这样的: 1.提取mRNA=》2. 建库测序=〉 3.拿到一定的原始数据量raw data【双端x测序深度x建库大小,比如测序深度20X,双端150bpillumina测序=》得到的数据就是2x150bpx20M=6G的数据量,但这个6G是碱基量,和硬盘占用空间的gigabytes(g)不一样哦】=》 4.质控过滤(具体参数设置可以参考一些发表的好文章他们的设置方式)=〉 5.clean data=》接下来兵分两路
研究物种是没有参考基因组的:从头组装转录本(de novo assembly)【工具包括:Trinity(可以预测更多的基因、转录本);SOANdenovo-Trans(更好检测高表达转录本);Oases(有效检测低表达基因以及长的亚型,N10-N50值高)】,然后再将reads比对到重装的转录本上,利用RSEM/Deseq2可以计算表达量
研究物种是有参的,参考序列可以选基因组或者转录本,然后用featureCounts(主推)/RSEM/htseq定量
关于featureCounts:http://bioinf.wehi.edu.au/featureCounts/
如果选基因组为参考,那么就要考虑到基因组含有的内含子和外显子。我们知道,转录的过程其实是把内含子去除了的,因此我们使用比对软件就要支持跨外显子拼接(exon-exon juction) 。最新的当属"Hisat2+Stringtie”,stringtie的merge算法会按照去掉内含子的基因组把所有样本的组装结果合并
如果选转录本为参考(前提:一般有基因组但不完整),那么转录本已经是去除内含子的序列了,因此计算压力就没那么大,比如常用的BWA、Bowtie2、STAR都可以; 或者可以用非比对定量软件Sailfish【它原理是利用参考序列和k-mer长度建立索引,然后将reads直接比对到转录本,然后采用EM(Expectation maximazation,最大期望)算法估计转录本与基因极大似然估计】
这样只能对已知的转录本、基因定量,不能预测新的
去向何方
表达量数据得到了,下面就要对感兴趣的表达差异进行探索,**这里面的思路就是:**我们利用一种试剂、毒素、药物处理后发现,处理后的和未处理的在某些表型出现了变化,而这种变化可能正是我们感兴趣的【比如就有这么“有(wu)趣(liao)”的导师扔给你两只老鼠🐭:“去,把其中一只给我处理了,做出个高分文章来”——听花花讲的】。
我们可能开始并不知道差异背后的机制是什么,但是可以通过探索一些有变化的基因来分析:参与何种生物途径的基因发生了变化,进而结合分类数据库GO、代谢通路数据库KEGG做一个推测,为下一步研究打个基础(这也是如今转录组只适合打辅助的原因,因为它只能给你推测)
差异分析三R包法宝:Limma、DESeq2、edgeR【特殊情况:无重复用GFOLD(命令行)】
源自芯片的金标准Limma:芯片数据普遍认为符合正态分布 ,而正态分布的群体一般就是用t检验(两个样本)或者方差分析(多个样本)。芯片数据量是很大的,limma据此而生,并且支持多重检验校正来控制假阳性
关于二项分布、泊松分布、正态分布的关系:
简单来讲,泊松分布,二项分布都是离散分布;正态分布是连续分布 http://hongyitong.github.io/2016/11/13/%E4%BA%8C%E9%A1%B9%E5%88%86%E5%B8%83%E3%80%81%E6%B3%8A%E6%9D%BE%E5%88%86%E5%B8%83%E3%80%81%E6%AD%A3%E6%80%81%E5%88%86%E5%B8%83/
另外,方差分析其实就是看组内方差显著性与组间方差的对比,据此判断不同组(也就是不同样本)之间差异是否存在
limma采用贝叶斯模型(Empirical Bayesian model),更新的limma-voom适配了转录组数据
RNA数据普遍认为符合泊松分布
DESeq2:采用负二项分布算法(negative binomial distribution),它的用法在https://master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html
RNA-seq中,技术误差是满足泊松分布的,因为期望和方差差不多。但是生物学重复之间的误差不能用泊松分布来描述,因为他的方差可能很大,所以要用负二项分布 https://www.cnblogs.com/leezx/p/6036806.html
它需要的数据是标准化之前的raw count(全部是整数),导入方式有四种:
function package framework output DESeq2 input function summarizeOverlaps GenomicAlignments R/Bioconductor SummarizedExperiment DESeqDataSet featureCounts Rsubread R/Bioconductor matrix DESeqDataSetFromMatrix tximport tximport R/Bioconductor list of matrices DESeqDataSetFromTximport htseq-count HTSeq Python files DESeqDataSetFromHTSeq 实际运行:其实可以一步搞定
#如果是自己构建矩阵的话 library(DESeq2) exprSet <- as.matrix(原始表达量矩阵)# 每列表示一个样本,每行表示一个基因,必要时可以每行加上基因名 condition <- factor(c("control","case1","case2")) #表型 dds<- DESeqDataSetFromMatrix(countData = exprSet,colData = data.frame(condition),design = ~condition) dds <- dds[ rowSums(counts(dds)) > CUT, ] #【选做,表示过滤一部分count值低于CUT数字的数据,count自定义】 dds2<-DESeq(dds) #开始运行 result <- results() #查看结果 mcols(result, use.names = TRUE) #可以看每一个结果的意义,利用其中的指标可以画图 summary(result) #看总体结果,多少上调,多少下调【一般认为foldChange绝对值 > 2以及p value < 0.05就是差异基因】其实这中间经历了三步计算:
estimateSizeFactors、estimateDispersons、nbinomWaldTest[选看:]DESeq2用自己的算法进行归一化: 第一步:
estimateSizeFactors计算每个样本的sizeFactor相关的过程是这样的:将原始表达量矩阵先进行log转换,然后计算每个基因在所有样本的均值rowMeans(log(raw_counts)),再将要计算的单个样本中每个基因的表达量减去前面的均值,再取中位数,就得到了sizeFactor; 第二步:有了每个样本的sizeFacor,再将样本原始表达量除以sizeFactor,就是归一化的表达量edgeR: 使用
DEGList读取表达矩阵=》 利用(count-per-million,CPM)严格过滤count值低的数据=〉calcNormFactors函数使用TMM算法对矩阵标准化=》 实验设计矩阵Design matrix,model.matrix(~0+group)=〉 估算离散值dispersionestimateDisp=》 构建比较矩阵makeContrasts、glmQLFTest=〉 提取差异基因decideTestsDGE、glmTreatlibrary(edgeR) exprSet <- as.matrix(原始表达量矩阵) condition <- factor(c("control","case1","case2")) #表型 dds <- DGEList(counts=exprSet, group = condition) # 利用count-per-million(CPM)标准化 keep <- rowSums(cpm(dds) > 0.5 ) >= 2table(dds) genelist.filted <- genelist[keep, ,keep.lib.sizes=FALSE]