056-一起来爬聚类树2
刘小泽写于18.11.22
今天看看主流软件MEGA的使用,我也是第一次接触这个软件,之前没做过任何聚类树。所以,没有基础的不用担心,边学边用边写就是了😉
什么是发育树?
长期的物种发展过程中,会由于基因和环境的影响导致自身发生进化或者退化,当然不管进化还是退化,都算作物种的演化,但是我们在平时说的时候喜欢说“进化树”、“系统发育树”而不是“演化树”。
系统发育树可以分为物种树(物种间关系)、分子树(根据DNA/RNA/protein等分子数据),这里要注意:单个分子的亲缘关系远近并不能直接反映物种的关系【因为如果换另一个分子做发育树,可能物种的亲缘关系就会发生变化】。总而言之,仅仅几个点并不能得到全部信息,想没想起来盲人摸象的故事?
上面说的几个点的意思就是:建树的分辨率问题,仅仅用一个或者几个基因建树,如果基因间亲缘关系近,那么就比较难分开;基因间亲缘关系太远,结果的同源性就太差。要想做的精细准确一些,将树分的开一些,就是需要提高分辨率,也就是在全基因组层面上提取SNP、InDel等信息进行建树。有两种方式(任选其一):
- 全基因组SNP建树:利用不同样品在全基因组水平上的SNP信息构建,包括了基因区和基因间区的信息,但不包括InDel信息
- 全基因组coregene集建树:利用不同样本在全基因水平上共有的保守单拷贝基因,包括了SNP与InDel,但不包括基因间区
系统发育树还可以根据拓扑关系分为 有根和无根树 有根树需要先找到所有样品的祖先,作为树的根(外群),其中所有样品与根的距离就可以反映物种演化的途径与时间长短;无根树就是把所有物种整合成几个大类(比如纲的级别),但并没有关于演化的信息

怎么看发育树?
树的样式多种多样,有长条的、圆圈的,有根的、无根的,那么怎么看其中包含的信息呢?
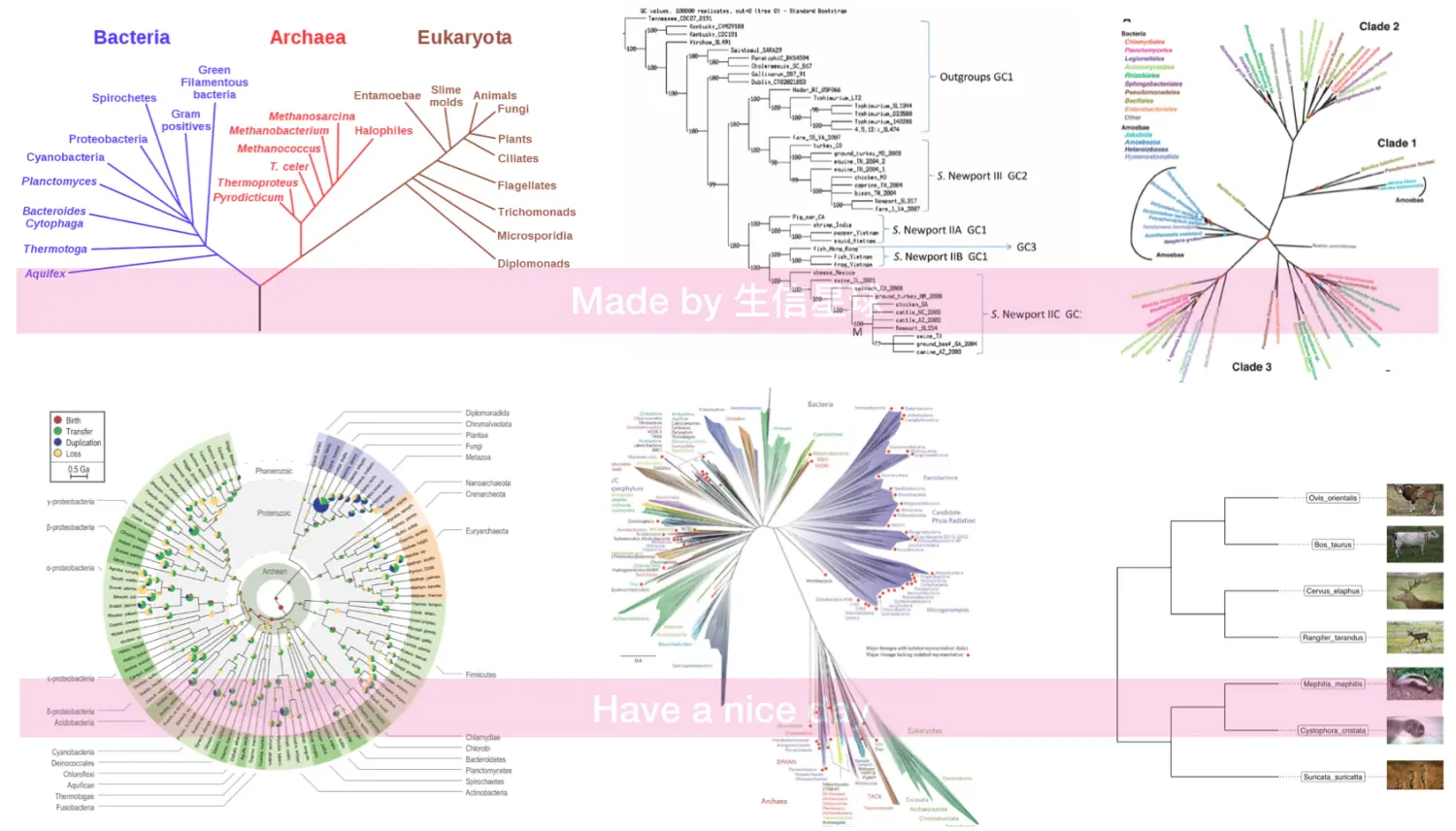
这个可能乍一看上去比较难理解,但是发育树已经深入了我们的生活,比如我所了解的广东地区村村有牌坊,有祠堂,其中保存着相当完整的家谱,同一代的叫“旁系”(如兄弟姐妹),上一代和下一代叫“直系”(如父母与子女)
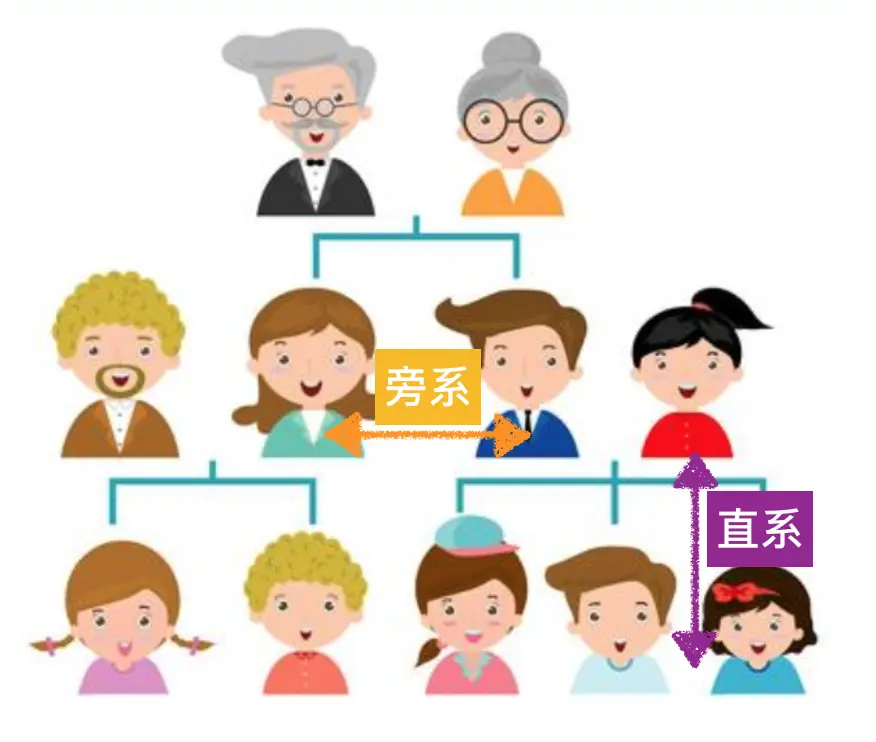
根据这个原理,目前很火的基因测序分析中的“祖源分析” 就是利用测序结果,帮你找到自己在系统发育树中的位置,这类分析美国流行的比较早,可能和他们是移民国家有关吧,多少代人生活在那里,却找不到自己的根,所以这种好奇心迫使他们开发了这种技术。另外,现在各个国家都在致力物种全基因组测序,想要尽可能地把存在的物种都测一遍,这个亲缘关系图谱的意义可就大了,不亚于2003年的人类基因图谱。因为一旦有了这个全物种图谱,回答当初那个**“我们从哪里来”**的问题就容易多了,可以解释我们是如何从单细胞演化到现在的顶级生物的。
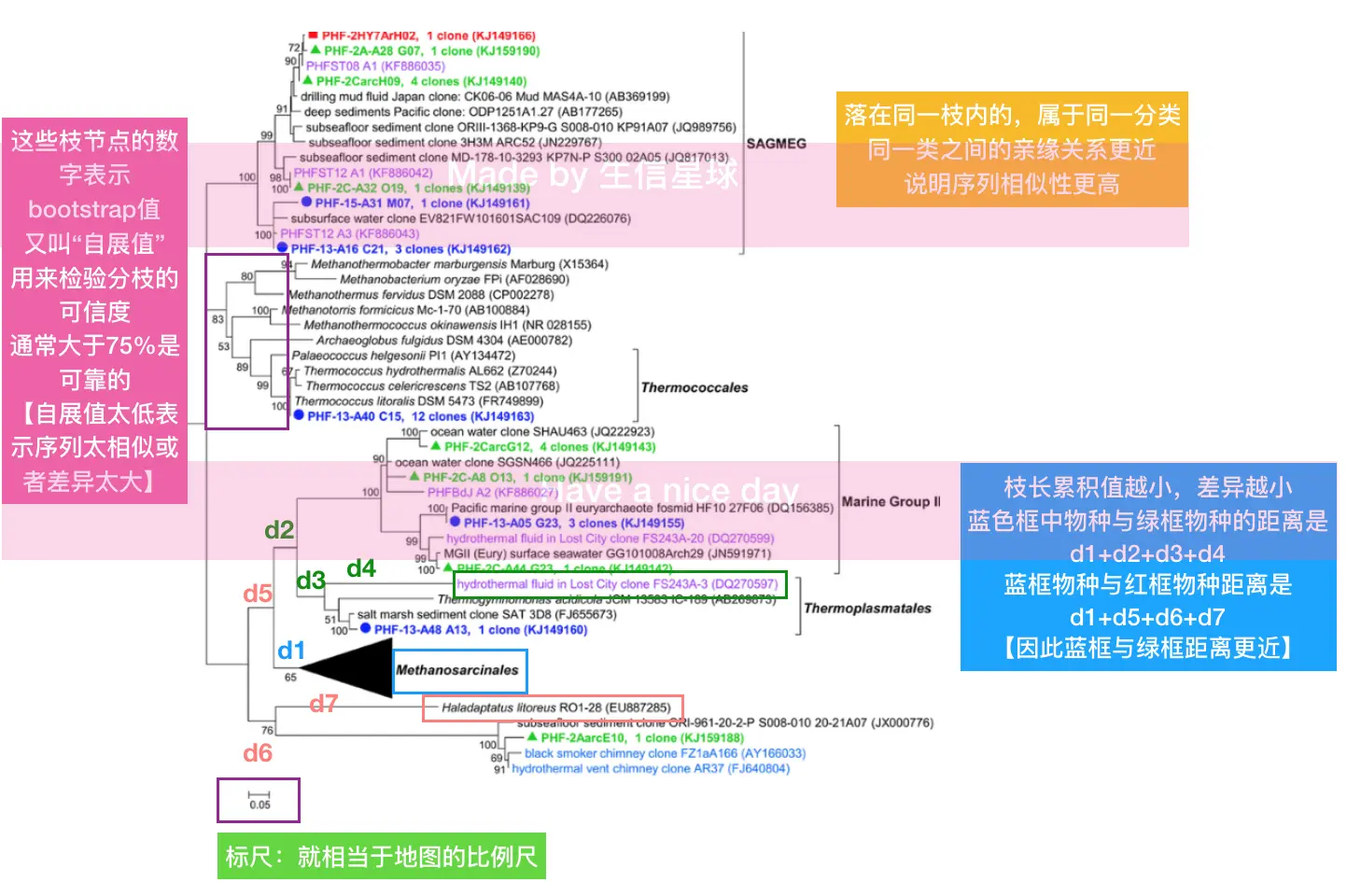
数据到树怎么做?
需要了解一下作图的算法,但是算法不是根本,它只是一种实现手段 关键在数据,我们画树不就是为了根据序列的关系反推样本之间真实的亲缘关系吗?那么如果真实的样本关系就是如此,不论用什么算法,做出来的结果也基本一致
如果,用了不同的算法,结果差别特别大,那么这时就应该想了:这不是算法的问题,数据本身有问题的可能性就比较大
聚类分析日常可见,比如商店、超市都会把商品分区分类,方便寻找。数据到树,本质就是利用无监督的机器学习手段,实现聚类分析【这里无监督的意思就是:在聚类分析之前,程序不清楚要聚成几类】只不过将序列进行聚类,靠的就是样本间显著差异的阈值点
常用的聚类
层次聚类
每个观测先自成一类,然后每次两个类再合并,直到所有的类被聚成一类
算法:单联动、全联动、平均联动(UPGMA)、质心 、Ward法
划分聚类
先制定类的数量K,然后将样本随机分成K类,再将这K类重新聚合
算法:K均值(K-means)、围绕中心划分(PAM)
这些算法先不用知道具体的意思,只要知道有这些东西就好了。然后需要知道它们的作用,是计算距离矩阵的
计算距离最简单的办法是用欧几里得距离(欧式距离)【根号下的差值平方和】,但是欧式距离适用于连续型变量,比如一堆数字;而我们要计算的是序列,属于名义型变量 ,所以不用欧式距离。
在层次聚类中,单联动是一类与另一类中点的最小距离;全联动是一类与另一类中点的最大距离;平均联动是一类与另一类中点的平均距离;质心是两类中质心间的距离;ward法是两个类之间所有变量的方差分析平方和
常用的方法
NJ邻接法(neighbour joining):计算速度快,适合序列相似度较高的序列;相似度很低,出现“长枝吸引(Long branch attraction, LBA)”【即:最相似的支系将优先被群组在一起,而古怪的支系(即具有长枝者)则被排除并被分成一群(来自:维基百科)】
UPGMA: 数据是连续型,如数字矩阵、差异表达矩阵
MP最大简约法 (Maximum parsimony):适用序列数目较少且差异较小,一般不用于远缘序列
ML最大似然法(Maximum likelihood):序列之间亲缘关系略远,不同的谱系演化速率差别较大
MEGA怎么用?
简介
这个MEGA应该是建树首选了,scholar数据显示经典款MEGA5引用接近37,000
MEGA是Molecular Evolutionary Genetics Analysis的缩写,第一款诞生于1993年,到目前发展到正式版 versionX (mac用户需要用公测版的X版本)。起初只有Win和Mac的图形界面,但是后来随着数据量越来越大,命令行版本的MEGA CC版本诞生
MEGA的官网:https://www.megasoftware.net/
下载版本X,官方声称两大优势:好看、并行运算
界面
先熟悉下顶部的工具栏【主要功能都在这】
ALIGN进行比对;DATA用来输入输出数据;MODULES计算相关模型;DISTANCE计算距离矩阵【核心步骤】;DIVERSITY计算多样性,也就是计算pi值,尤其是动植物群体选择压力分析中可以用到【pi值是计算两两序列之间差异度,再求平均值】;POLYGENY画树功能;USER TREE用于分析和编辑画好的树;ANCESTER寻找祖源序列
再看下底部信息栏
有帮助信息、参考数据、引用信息等,其中帮助信息可以好好看看,非常详细
点击EXAMPLES会出来对话框,参考数据随软件一起安装进来,直接使用
https://www.megasoftware.net/examples 官网也列出了示例数据的含义
Mega自带了文本编辑器功能,File=》Edit a text file,然后打开示例数据中的hsp20.fas (官网对这个数据的解释是:Unaligned HSP20 sequence data for four species)

这个是标准的fasta格式数据,>开头,然后紧接物种名或者样本名(这部分要先设置清楚,因为建树就是显示的这部分ID),然后空格
另外再看一个meg格式的文件Drosophila_Adh.meg这是一个多序列比对的Mega自定义文件。特点是:第一行是#mega 作为标志,然后包括Title、Format、Description等信息,并且以!开头,接下来就是正式的序列比对信息

流程
- FASTA序列输入、比对【可以将多个物种的序列放入同一个文件夹】
- 多序列比对生成meg/mas格式的比对结果文件
- 导入画树功能中,计算距离矩阵
- 生成Newick格式的树文件
第一+二步 序列的输入、比对、保存
DATA=>Open a file => 选择hsp20.fas=>提示align or analyze (其中analyze是利用自带的文本编辑器进行编辑)=》align=〉得到多序列比对图形结果
关于多序列比对:
- 三条以上DNA/RNA/protein序列
- 输入序列有同源区域,不可以随便用几条序列放在一起进行比对
- 序列不要太长,因为多序列比对的过程是两两比较的过程,多一个样本,计算量就多一倍
- 序列方向保持一致,多序列比对不像全局比对或局部比对可以正反、交叉比对,如果存在一条方向与其他相反,那么比对是失败的
- 注意单条的离群序列,可以删除掉
进行比对:
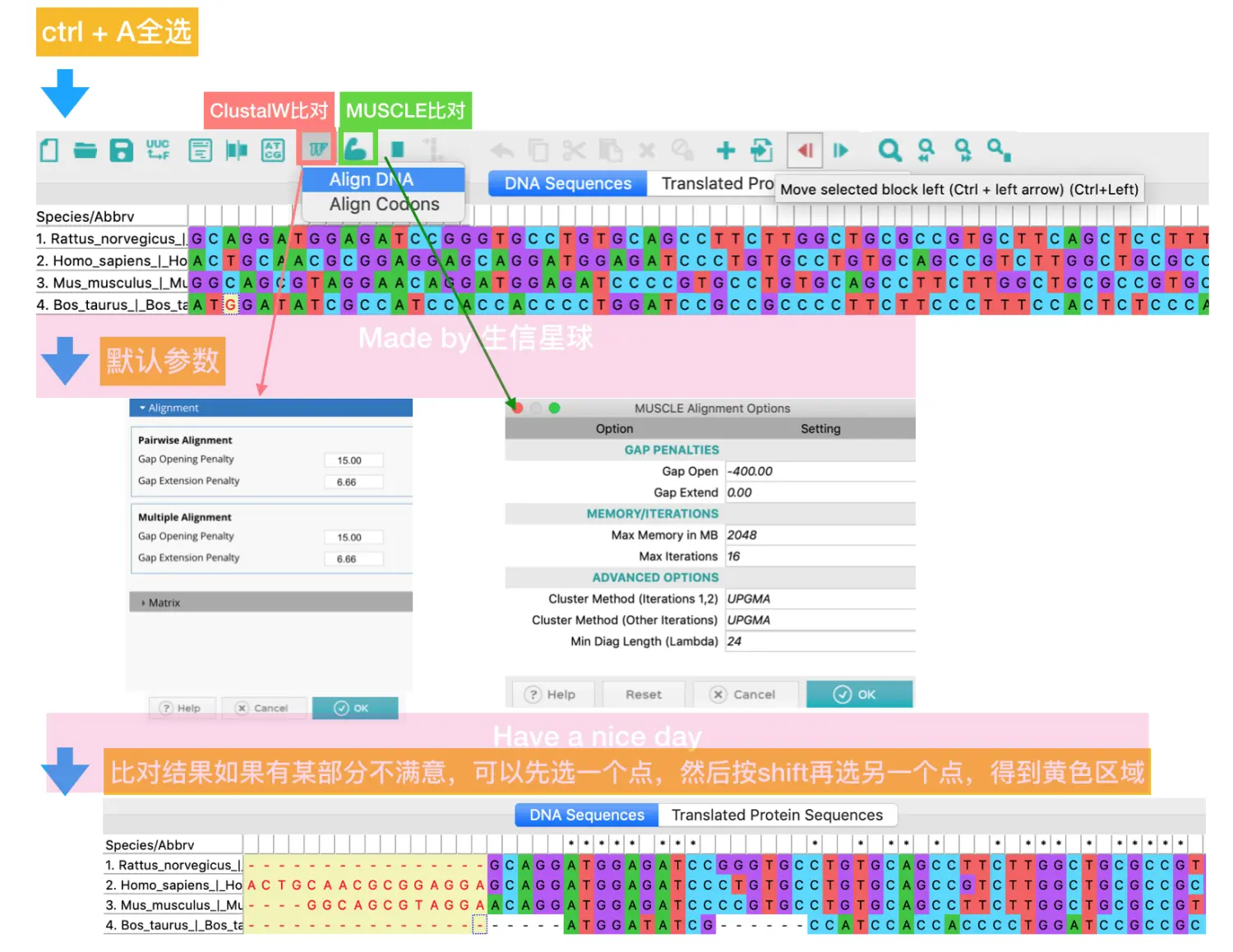
最后保存比对结果,默认格式是mas,另外可以在Data => Export alignment中选择其他格式
另外,可以将DNA序列翻译成氨基酸序列再进行比对【一般认为氨基酸具有更好的保守性】,返回多条序列显示的界面,还是ctrl + A全选,然后选择第4个图标,会提示“是否将选中区域进行翻译”=》选择yes=〉然后提示使用标准的密码子表(当然也可以自定义)=》此处还是yes=〉得到翻译后的氨基酸序列=》用上面的方法继续比对
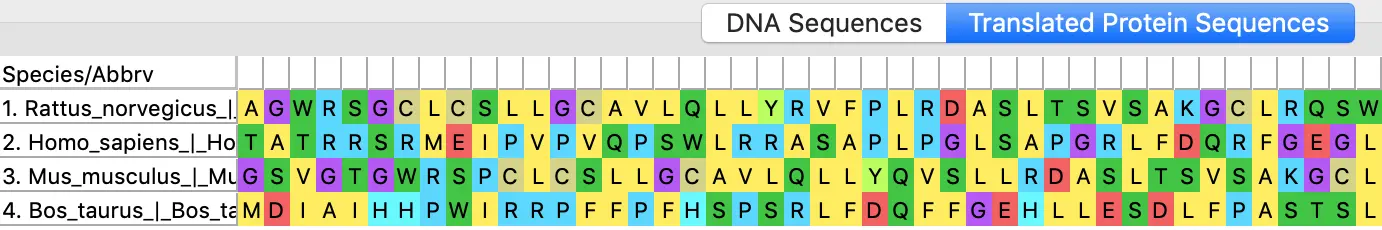
Cool Tip: 在线下载序列并比对
ALIGN =〉Edit/ Build a new alignment (新建一个空的比对环境) =》选择类型DNA/protein =〉菜单栏找到Web =》Query Genbank就会打开NCBI 核酸数据库=〉输入想要的序列名称=》在左侧筛选物种,过滤出部分序列 =〉上方Summary中的FASTA(text) =》程序会把这些序列都打印出来=〉Add to Alignment
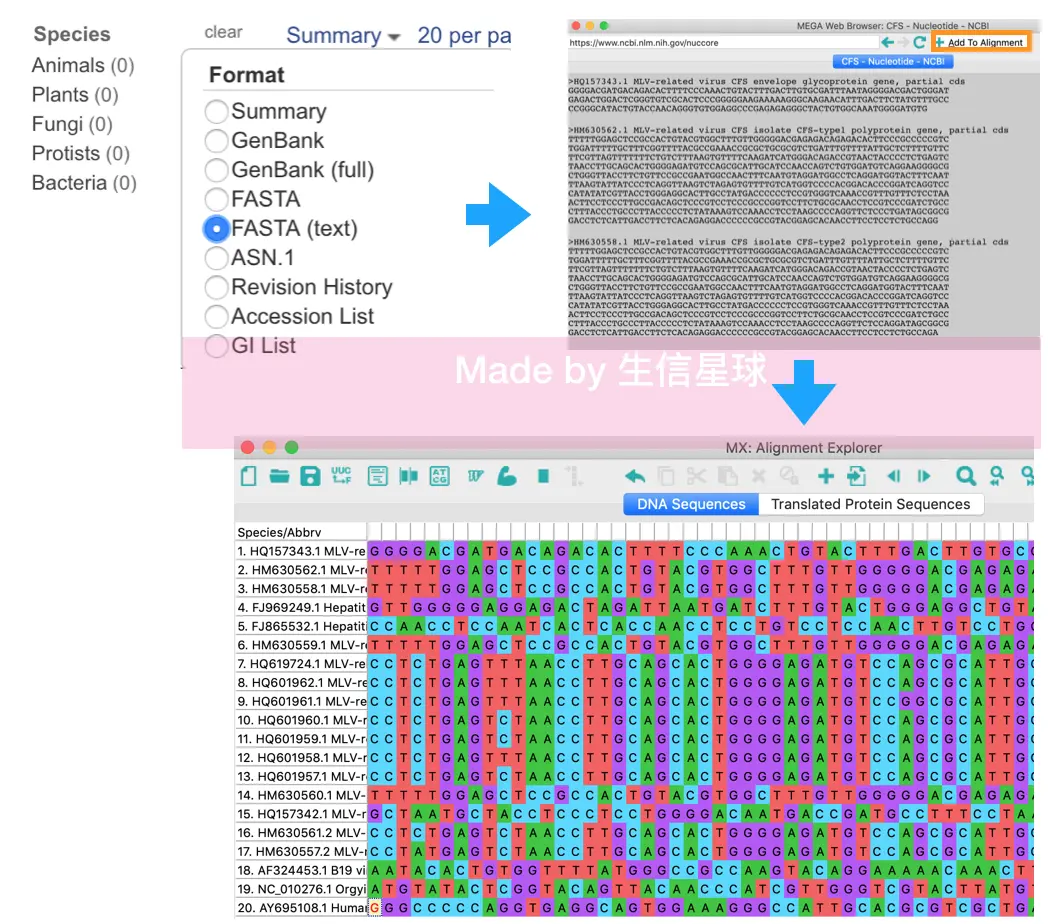
第三步 计算距离矩阵
假如现在有一个比对好的meg文件
通过DATA打开,这里以Drosophila_Adh.meg为例,然后主界面会出现两个图标,左边的是序列比对(TA),右边是关闭当前文件(Close Data)
点击左边图标=>查看序列比对文件=>先看下工具栏
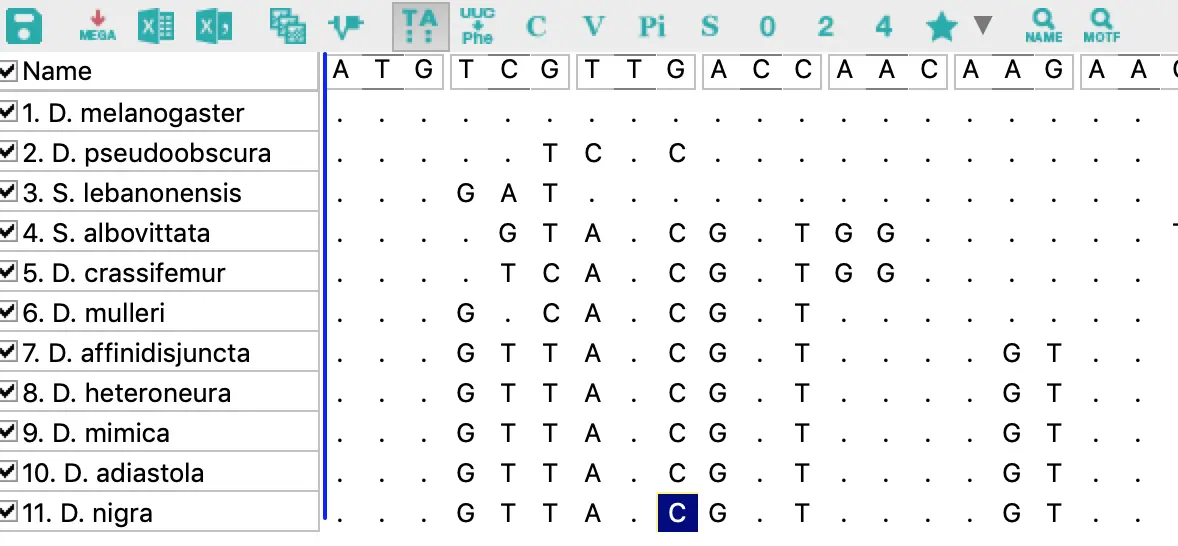
第1个图标可以设置保存格式;
第5个图标可以对序列进行分组,点击New创建分组;
第6个图标是编辑和选择基因或功能域;
第7个图标是显示或隐藏相同的序列位点;
第8个图标是核酸与氨基酸的互换(这个功能在平时的序列转换也可以用到);
然后C、V、Pi、S、0、2、4都是高亮显示序列【其中C是高亮全部一致的部分;V是存在变化的部分;其他的可以在帮助文档中搜索“highlight”】
另外在菜单栏中,还有数值统计Statistics,统计核酸组成、使用的密码子,并导出txt文本
然后使用DISTANCE
结果表中的数字表示样品中两两亲缘关系(即相关性系数)。如果一种方法得到的相关性系数比较小,可能是计算方法问题(可以换一种再试试),也有可能是同属不同种的生物,还有可能是不同属的生物,不太相关也比较正常
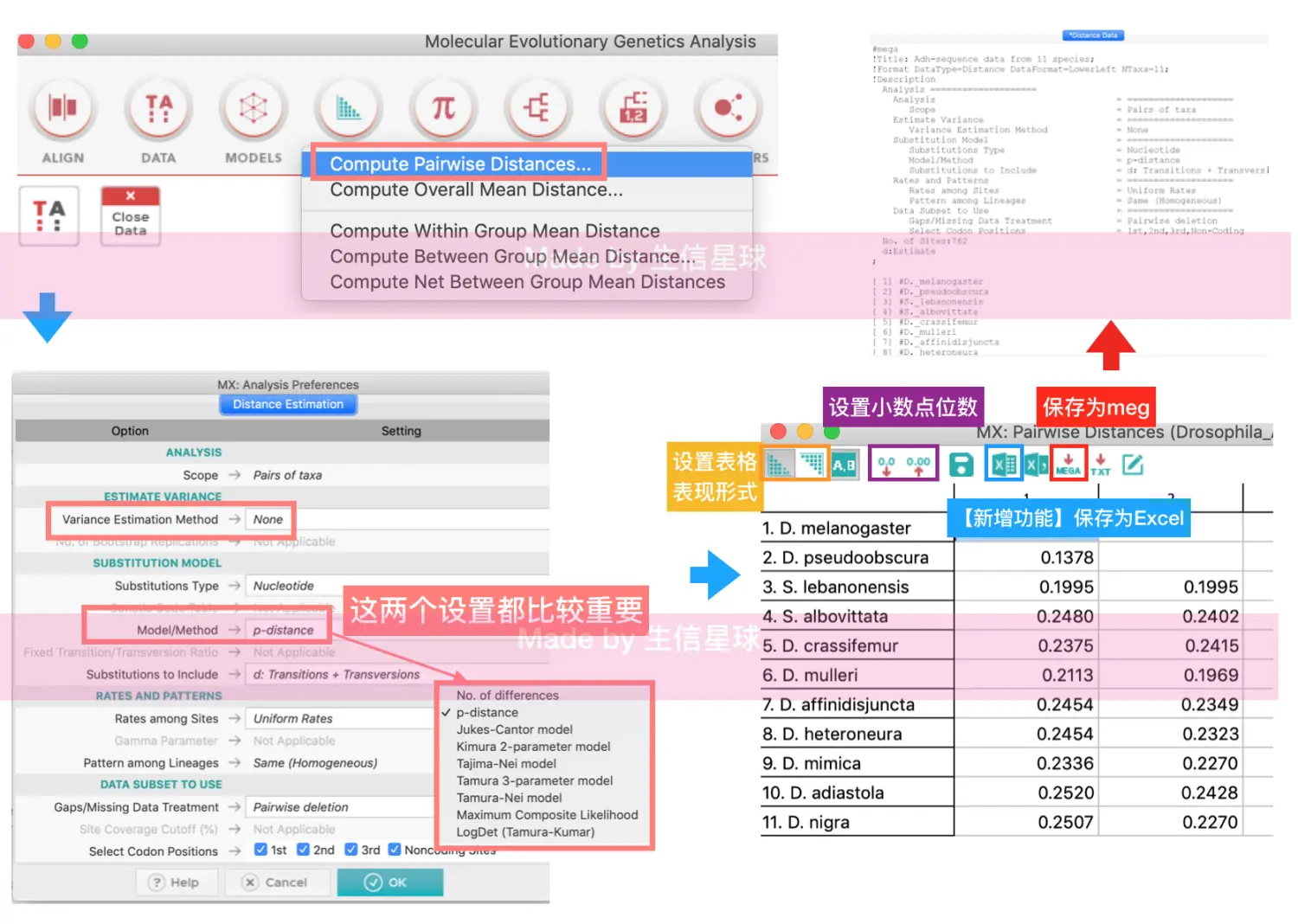
除了保存为meg、xls,还可以设置保存格式为csv,这样直接导入R中,用hclust也可以聚类
第四步 建树
其实构建距离矩阵后,可以直接建树,这里分步说明只是为加深对每步印象
打开距离矩阵meg文件,然后选择PHYLOGENY
先用NJ算法得到一个树,得到两种树:original tree和Bootstrap consencus tree , 在original tree中有枝长、bootstrap值、标尺
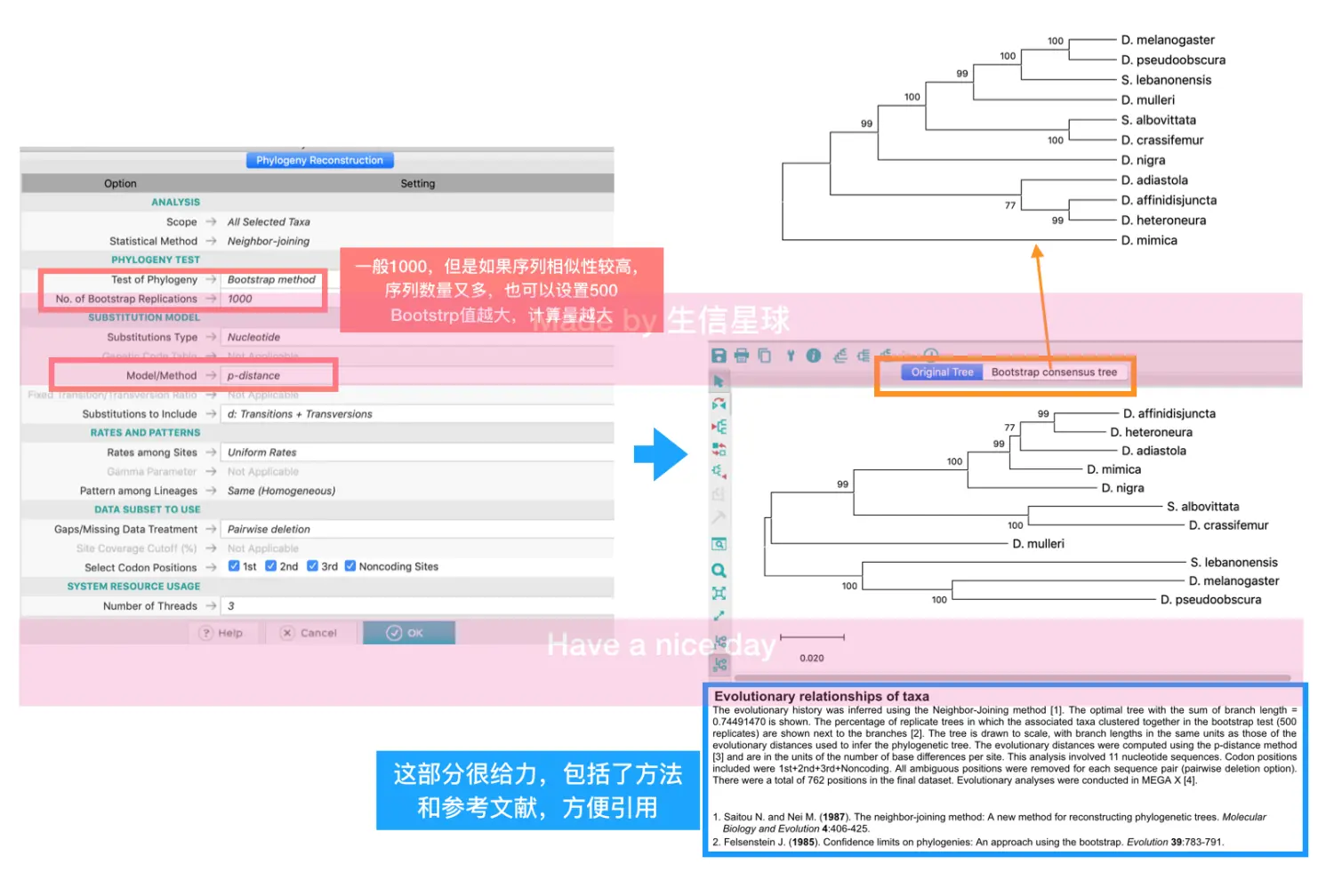
构建完还可以优化结构,可以在顶部的工具栏中有三个聚类树形状的按钮。第一按钮可以设置圆形或者放射状等形状,第二个按钮可以让文字对齐(使用Toggle scaling),第三个按钮可以对树进行过滤。
在左侧工具栏中设置显示node ID、node bootstrap值、枝长值
想要自定义颜色、字体等,可以使用顶部工具栏的小扳手图标,它还可以进行枝干的设置、进行缩放。其中Tree选项是对整个树进行设置,包括枝长、宽度等;Branches选项用来为枝干加数值,修改线条粗细;Labels 设置节点的形状、颜色以及taxon name的字体、颜色;scale设置标尺;cutoff过滤不满足阈值的枝
第五步 发育树美化
使用iTol (Interactive Tree of Life)进行美化,是一个在线操作聚类树的工具,除了系统发育树,还有一些关系树等,另外除了树,还可以添加热图、柱状图、蛋白结构域等。需要注册使用,使用时先新建一个workspace,然后将netwick格式的树文件上传【tips:一般比较好看的树样本数量在30-150之间】之后所有的设置都是立等可见的,官方文档也十分详细