078-为什么要搞全基因组测序|测序了然后呢
刘小泽写于19.1.14
随着测序技术的不断升级优化,读长越来越长,某些基因组比较小的细菌可以实现从头测到尾,那么为什么我们要获得全基因组信息?它能为我们提供什么帮助呢?
认知的进化
首先对于生物这个词汇,我们的认知水平是在不断刷新的,从开始的生态学角度了解生物的形态结构、种群群落组成到生物的生理生化过程研究(例如物质代谢、能量流动),有了基因组后,我们就可以从基因组层面上对基因功能进行注释,然后比较不同生物的基因组差异,看看哪些生物的基因组特征值得被研究。总而言之,我们想搞明白我们从哪里来,能到哪里去。
基因组层面能分析些啥
这么高大上的词汇背后肯定有大量的分析要点,否则不用这么费时费力去取样、测序。另外,不管使用什么测序手段(Illumina、PacBio、IonTorrent等),最后得到的结果用处都差不多,大体上分为:结构基因组学、功能基因组学、比较基因组学(来自百度百科),感觉这么说还是不太明白。
想想我们做基因组不还是为了更好地去了解这个物种吗?
- 那么首先,要对这个物种本身达成一定的认知,比如基因组上哪些位置是基因?基因的功能都是哪些?与该物种表现出来的特有的功能相关的基因是哪些?另外除了编码区域,还有哪些区域是非编码RNA?哪些是重复序列?哪些编码比较特殊的基因元件?
- 除了分析自身,还可以与其他物种比较 ,发现它们之间的差别(包括单碱基水平变化:转换、颠换、插入、缺失;染色体水平变化:倒位、易位、插入、缺失),找到亲缘关系远近
- 找到基因组上的差异后,可以再和表型信息进行关联分析
举个例子:人贵在有自知之明,那么我们如何做到自知呢? 首先最了解自己的人就是本体啦(包括自己适合做什么,有什么兴趣爱好,对什么领域感兴趣)=》物种本身认知,然后自己可以再和其他人比较(看看哪些地方做的还有所欠缺,哪些地方值得发扬)=》其他物种比较。明白差异后,我们可以有的放矢,去寻找和自己爱好相关的工作=》差异与表型关联
基因预测
一般有两种方法:
- 和已知近缘物种基因集进行同源序列比对,筛选出同源比对区域,作为基因(就是利用已知的信息去预测未知)
- 从头预测:利用软件对物种的基因组直接进行预测(如果分析的序列有明显的特征,如:基因的编码区CDS与开放阅读框ORF、核糖体RNA的保守域、转运RNA的倒三叶草结构,就可以用软件识别结构并预测)
两种方法比较
从头预测:不需要同源参考基因序列,直接可以进行预测,适用于新发现的物种(因为没有足够的已知信息,因此需要先构建训练集【训练集:软件先对基因组的特征做一个调查了解】)
序列比对:找的基因是已知发表过的,结果更加准确,但是毕竟是近缘物种,不可能序列区域一致,因此可能同源区不含有某个基因或者有一段非同源区域恰好含有特征基因,这样就会漏掉一些
开放阅读框(Open reading frame,ORF)
从5’端开始翻译的其实密码子(ATG)到终止密码子(TTA、TAG、TGA)的蛋白编码序列。预测之前我们是不知道DNA双链中的哪一条链是编码链,也不清楚准确的翻译起始位置,但是知道的是:正负两条链每条都有三种可能的ORF,两条链共6种。于是我们就是利用这6种可能的ORF找到一个正确的,然后根据这个ORF得到氨基酸序列,最后预测出来蛋白产物
补充:不是所有的ORF都叫CDS
CDS,是编码一段蛋白产物的序列;ORF是理论上的氨基酸编码区;CDS一定属于ORF,当然可能包括许多个ORF,但是每个ORF不一定都是CDS。 ORF的识别是证明一个新的DNA序列为特定的蛋白质编码基因的部分或全部的先决条件。
原核生物-软件
基于HMM(隐马可夫模型)glimmer3:https://ccb.jhu.edu/software/glimmer/
Prodical:https://github.com/hyattpd/Prodigal
GeneMark:http://exon.gatech.edu/GeneMark/
相对简单,用自身的基因组作训练集即可
根据不同的物种,选择适合的密码子表 https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi
真核生物-软件
复杂的地方在于:ORF不仅包含编码蛋白的外显子(exon)还有内含子(intron),内含子将ORF分割成许多个小片段,导致ORF的长度变化范围很大。但是, 真核生物的外显子与内含子连接基本满足GT-AG规律(即:内含子序列的5’端起始的两个核苷酸总是GT,3‘端最后的两个核苷酸总是AG,5'-GT...AG-3' )
利用Augustus(http://augustus.gobics.de/) 包括人、大型哺乳动物、植物、鸟类、真菌基因组等
训练集:http://augustus.gobics.de/datasets/
除了基因预测,还可以用于从头预测,加入cDNA和EST序列,辅助提高预测准确度
一般可以选择多个工具进行预测,因为预测是非常关键的一步,会影响到下游基因功能注释部分,发表后别人还会以此来进行研究,因此,对于一个新的基因组,预测错误会产生“蝴蝶效应”
刘小泽写于19.1.16
看一下关于基因功能注释的前因后果
基因功能注释
背景
得到一个基因集以后,需要知道基因有哪些功能,参与哪些生物过程,只有理解了基因的功能以后,才能联系起来基因型与表型。
继人类基因组计划(HGP)完成以后,找到了2万多个编码蛋白基因,但是基因功能还未知,然后又有了ENCODE(Encyclopedia of DNA Elements)计划,找到了400万个基因开关,平均一个基因有约200个开关,这是人类既有共性又有个性的基础。ENCODE计划使人类基因组不再是个”空壳“ https://www.encodeproject.org/
关于ENCODE计划:https://kknews.cc/science/pq2nx8.html

简而言之,功能注释就是预测蛋白序列的功能,是最基本的分析之一
功能注释应用场景
- 从头拼接的基因组并做了结构注释,知道了哪些地方是外显子,哪些是内含子,接下来就是功能注释,预测每一条基因编码什么蛋白,并且蛋白是什么功能
- 无参转录组需要从头拼接转录本,拼接的转录本功能需要做注释
- 得到了差异表达基因,想做下富集分析,就必须要了解每个基因对应哪个GO分类,也是需要进行功能注释
原理
基因不同于蛋白,不能通过结构来预测功能,只能通过与已知基因功能数据库的比对去推测。一般的数据库包括了两部分内容:一是基因序列(核酸+氨基酸)FASTA格式;二是基因功能信息(可以写到FASTA的ID行中或者单独放在一个文件中)
一般采用氨基酸序列与数据库进行相似性比对,比对结果去数据库中进行过滤
这里看到,基因功能注释主要依赖数据库,如果数据库中没有这个基因,那么就无法注释。更可怕的是,数据库中有错误,就会进行错误注释
比对的结果并不是百分之百完全比对的,那么怎么判断氨基酸序列和数据库的关系呢?比对到多少才能被接受?这里需要考虑比对长度、比对分值、identity值等,过滤掉一部分人为认定不满足同源关系的序列。但是又有一个问题,不同区域的基因会发生不同程度的突变,如果仅设置一个值进行过滤——”一刀切“,这个结果还是有待优化
另外,如果结果提示:Selenocysteine (U) at position ** replaced by X 说明U氨基酸被替代成了X(当然并不是错误,可以忽略),因为在blastp/tblastn的打分矩阵中不存在U和- 这两个字符,替换成任意字符X就可以任意打分
https://www.biostars.org/p/111143/
基本流程
如果手头仅仅有几条蛋白序列想做下功能注释,那么直接甩给uniprot/ncbi在线blast比对就可以了,但是我们这里说的情况是成千上万条基因,肯定不能在线提交,那么怎么办?
要进行大量蛋白序列的功能注释,需要包括:同源注释、功能分类
同源注释
基于相似性的注释:就是将要研究的序列与蛋白数据库进行比对,将数据库中比对相似性高的蛋白序列可以作为研究序列的功能,常用的是Nr、Uniprot数据库 ,常用软件是blast和 diamond 【其中,blast速度很慢,比对几万条序列可能好几天甚至一周;diamond也是基于blast但速度最快达到blast的两万倍,准确性差不多,因此一般就使用diamond就好】
blast是基于动态规划算法,就是将每个位点都进行比对,比对上就得分,比对失败就罚分。从准确性讲是不错的,但是这个方法对于背后的生物学特性欠缺考虑。因为不是每个氨基酸都是一样重要的,对于某些抗性基因或者转录因子,真正起作用的往往是一些保守结构域
基于结构域的注释:Pfam(https://pfam.xfam.org/)数据库中有各种基因家族的保守域模型,可以用HMMER软件将研究序列与数据库中的模型进行比对,如果序列上存在某个结构域,那么推测序列含有该结构域功能;另外Interpro(https://www.ebi.ac.uk/interpro/)是一个综合数据库,使用interproscan软件比对
做完同源注释,就知道了研究的序列和数据库中的哪个蛋白最相似,我们主要利用了nr、uniprot、pfam、interpro这些蛋白数据库,它们又和下游的GO、KEGG、COG等分类数据库有关联,然后就能知道研究的蛋白属于哪个GO分类,哪个Pathway,哪个基因家族,就是功能分类
功能分类
只了解单个基因的功能是不够的,因为基因间是相互作用、协同完成生物功能的,所有需要进行分类,这就是在RNA-seq中得到差异表达基因后做的功能分类(GO)和富集分析(KEGG)过程,看看基因是不是协同完成某一个生物过程,它的原理与功能注释相似,也是利用已有的分类去推测未知的分类
小Tip:功能注释相当于一个过滤筛。GO 注释=》粗筛;KEGG=》细筛,例如:某一个蛋白,GO 只能将它注释到与细胞凋亡有关;而 KEGG 则可以将它注释到细胞凋亡通路中的某一个环节

例如COG数据库(Cluster of Orthologous Groups of proteins, https://www.ncbi.nlm.nih.gov/COG/)是细菌、藻类和真核生物的21个完整基因组的编码蛋白,根据系统进化关系分类得来的。
ftp://ftp.ncbi.nih.gov/pub/COG/COG 数据库还是2003年的,所以做出来的东西,看看就好了
# 下载数据库数据
$wget ftp://ftp.ncbi.nih.gov/pub/COG/COG/myva
$wget ftp://ftp.ncbi.nih.gov/pub/COG/COG/fun.txt
$wget ftp://ftp.ncbi.nih.gov/pub/COG/COG/whog
# 清洗COG数据库(只挑有注释的那些序列)
# https://gist.github.com/Buttonwood/96f9a9ef8159ca111a69
$cog_db_clean.pl -myva myva whog > cog_clean.fa
# blast+比对
$makeblastdb -dbtype prot -in cog_clean.fa
$blastp -query yourdata.fa -db cog_clean.fa -e 1e-4 -out blast.out -outfmt 7 -num_threads 10 -seg no
#整理结果 https://github.com/kodayu/blog_html/blob/master/blast_cog.py
$blast_cog.py blast.out fun.txt whog out
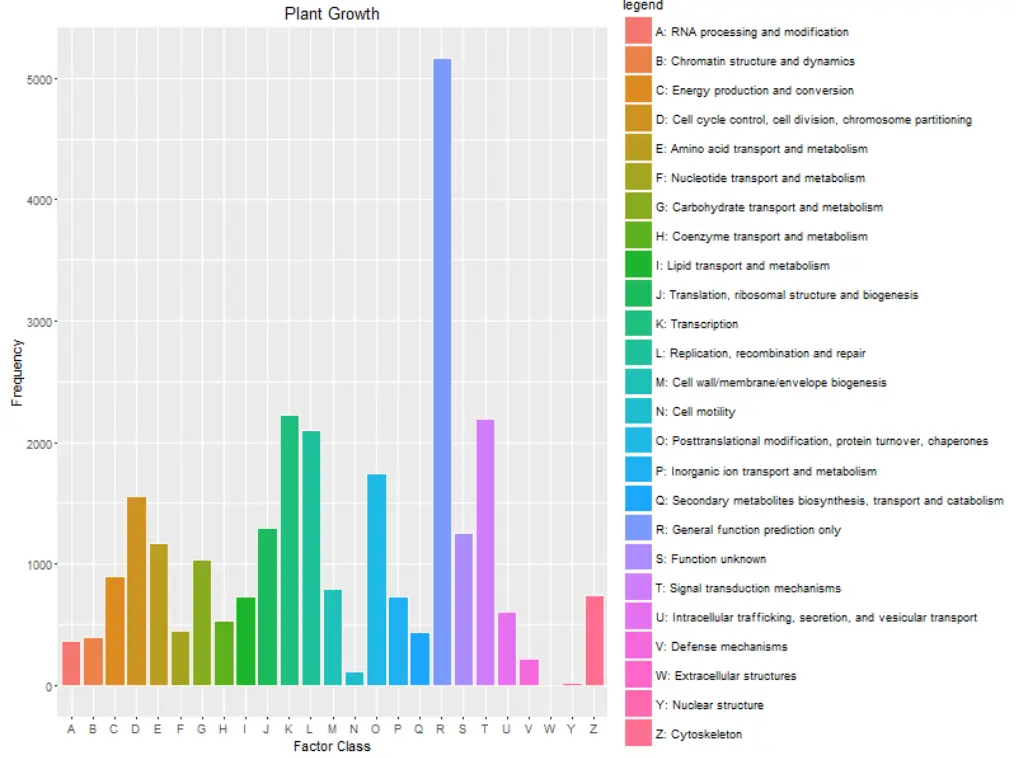
参考:http://yk.cancerbio.info/myblog/cog%E5%88%86%E6%9E%90/
又例如GO数据库 ,其中每个注释都是对基因产物的描述,有特定的分子功能(MF),涉及到特定的生物过程(BP),作用在特定的细胞组分(CC)。它把所有候选的靶基因向GO的各个term进行映射,然后计算映射到每个term的靶基因数量,在整个参考基因背景中利用超几何分布检验,选出候选靶标基因中显著富集的GOterm
再例如KEGG数据库,关于生物化学途径的描述,许多活细胞的功能不能仅仅依赖于单个基因,它将基因组信息与更高一级的功能信息结合;另外它可以将基因组中的许多基因利用细胞内分子互作网络联系起来,通过通路或者复合物来展示更高级的生物学功能
功能注释整合的流程
刘小泽写于19.1.16
功能注释这么繁琐吗,有没有整合起来的流程?
之前看到,同源注释和功能分类都有各自的数据库和软件,并且同源注释还有两种方式,一步步操作会比较繁琐,因此就有软件看到了这个痛点,开发了新的整合式流程
最有名的blast2go:https://www.blast2go.com/ 顾名思义,它是从blast直接给你GO分类结果,一步到位,但是目前专业版开始收费最便宜1600欧元/license,但提供一星期的试用。那么专业版好在哪里,官网也做了说明,主要还是强调:数据库更新快,分析结果更准确,还会送一个AWS云计算(https://www.blast2go.com/blast2go-pro/cloudblast 不需要用户自己安装数据库,及时更新,加速等等)
当然,基于blast软件,即使加持云计算优化,速度还是个问题;另外配置过程非常复杂
Trinotate:做过无参转录组的小伙伴都应该用过拼接转录本的Trinity吧,同样,这款软件也是Trinity团队为功能注释开发的(他们真的想从头到尾全实现了啊),这个软件的程序全是基于perl,所以不了解perl就不知道怎么解决报错
interpro+interproscan:部署也是个麻烦事,并且只支持基于结构域的注释,对KEGG、GO等数据库关联性不是很好
eggnog-mapper:2016年基因家族数据库EggNOG v4.5.1开始进入大家的视野,这个团队为数据库开发了相应的注释软件。目前已开发出5.0版本(http://eggnog5.embl.de/#/app/home) 支持了5,090个物种,比上一个版本4.5.1高出一倍不止。另外支持的Orthologous Groups也有4.5.1的19w疯狂升级到440W,其中每个Orthologous Group的蛋白也都关联了GO、KEGG数据库
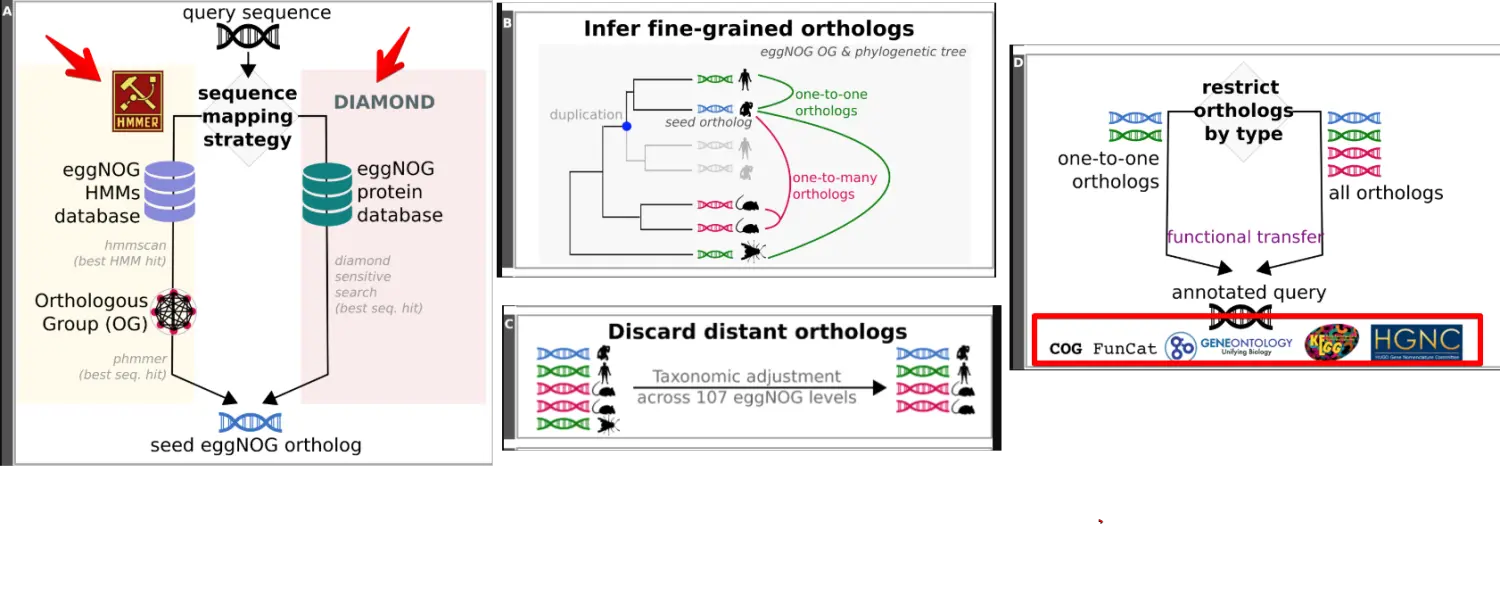
看来eggnog-mapper将是使用重点,那么它的设计原理是什么?
首先是比对,将要研究的序列与Eggnog中的蛋白序列进行比对,这个过程可以是基于相似性(DIAMOND),也可以是基于结构域(HMMER);
比对完以后就知道了我们提交的序列与哪个Orthologous Group最相似,并且还能知道与什么物种相似度最高,帮助确定注释的物种(真核还是原核,植物还是动物);
然后通过数据库之间关联进行COG、GO、Pathway的功能分类
安装eggnog-mapper
下载地址:https://github.com/jhcepas/eggnog-mapper/releases
另外还有详细的帮助文档:https://github.com/jhcepas/eggnog-mapper/wiki/Installation
或者直接conda安装eggnog-mapper
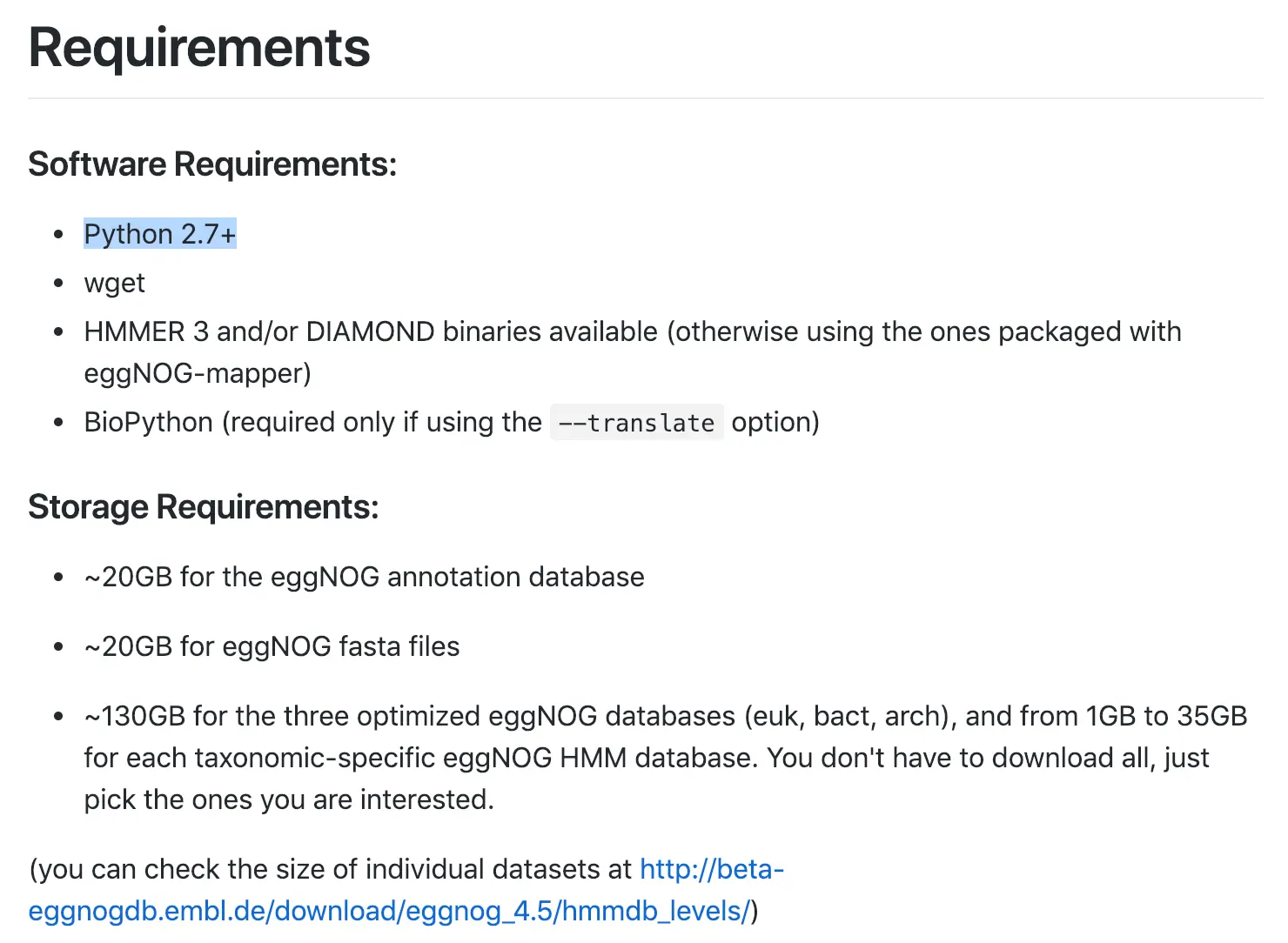
可以看到:这个软件基于python2.7,需要200G左右的硬盘空间,并且对于真核生物需要90GB的内存,细菌大约32G内存,古细菌10G内存 (因为使用快速模式的话,会把数据库一次性加到内存中)
下载数据库
它构建了107个各个层级的物种注释数据库
在这里http://eggnogdb.embl.de/#/app/downloads可以下载特有的数据库
当然也可以用download_eggnog_data.py euk bact arch viruses -y下载真核、细菌、古细菌、病毒(euk、bact、arch、viruses)的数据库
【如果是用conda安装的,命令可以直接使用;但自己手动安装的,需要把软件放入环境变量,并且还需要在开头加一个python2表示用python2进行编译】
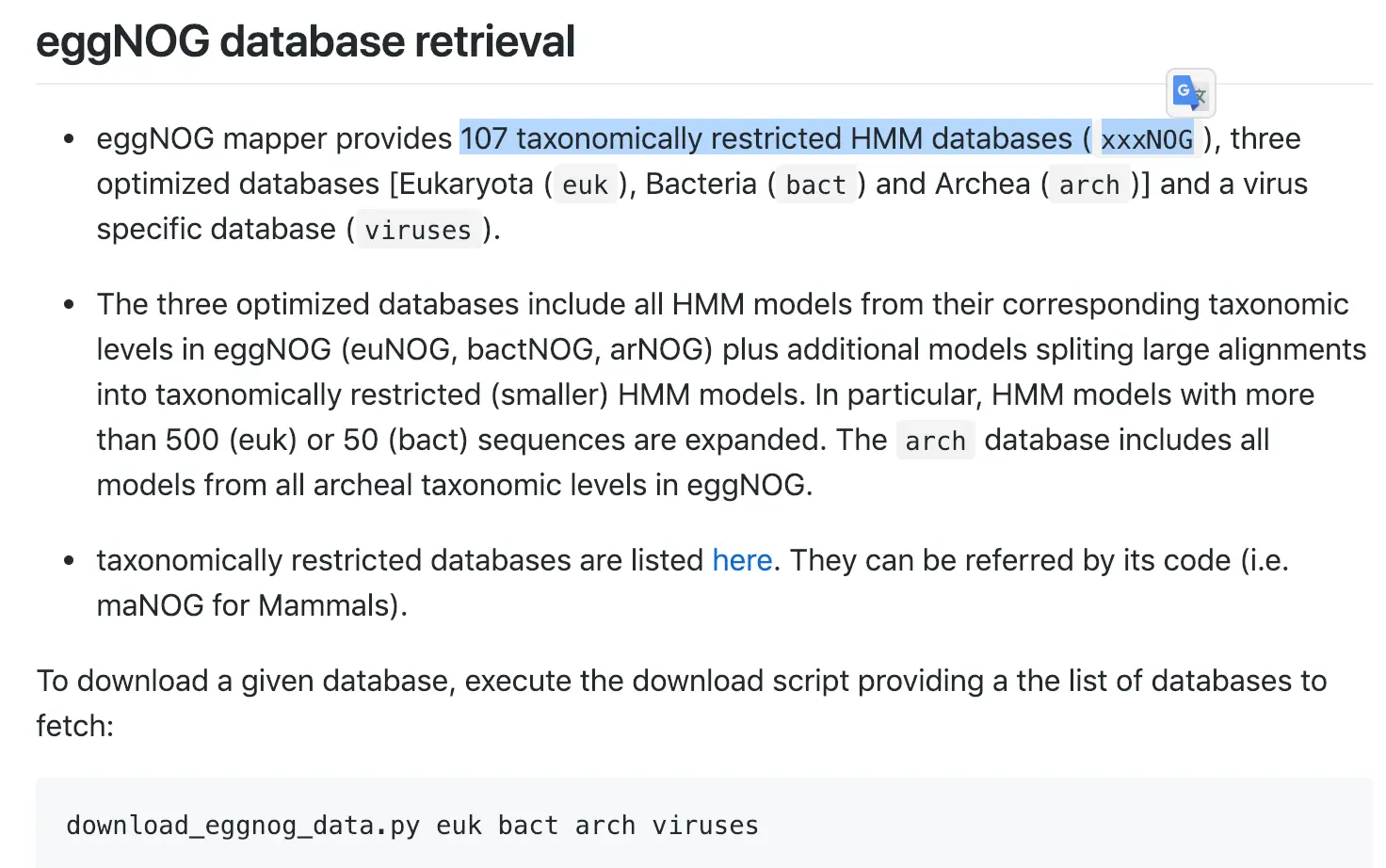
下载完成看看,主要包括以下内容:
其中OG_fasta存储fasta文件;eggnog.db存储eggnog数据库中的蛋白序列ID与GO、KEGG等其他分类数据库的对应关系; eggnog_proteins.dmnd是diamond的数据库;hmmdb_levels这个文件夹存储HMMER的数据库
OG_fasta/ eggnog.db hmmdb_levels/
OG_fasta.tar.gz eggnog_proteins.dmnd og2level.tsv.gz
配置好了,然后看看怎么用
如果使用conda安装,直接激活安装的环境,然后emapper.py -h 查看帮助文档
-m {hmmer,diamond} Default:hmmer #设置模式,默认是hmmer
-i Input FASTA file containing query sequences #输入的文件FASTA
-d specify the target database #指定数据库(euk,bact,arch,viruses从中选择一个)
-o base name for output files #设置输出文件前缀
--usemem #[可选]针对hmmer模式(一次性将数据读入内存,快速处理,真核生物推荐90G以上内存服务器使用)
首先看比对模式有两种:diamond、hmmer
diamond(对于大量的序列比如多于1000条)
$ cat >diamond.sh emapper.py -m diamond \ -i test.fa \ -o diamond # 不需要指定数据库 -dhmmer(少量序列比对少于1000条)
$ cat >hmmer.sh emapper.py -m hmmer \ -i test.fa \ -d euk \ #(bact/arch/viruses) -o hmm-euk \ --usemem
实际运行下病毒的测试数据
这里选用的是Zaire ebolavirus病毒蛋白数据,因为很小所以比较好操作
数据下载
esearch -db protein -query PRJNA257197 | efetch -format=fasta > protein.fa
先构建小数据,随机选100条蛋白序列
$ seqtk sample protein.fa 100 > test.fa
# cat >hmmer.sh
$ emapper.py -m hmmer \
-i test.fa \
-d viruses \
-o hmm-vir \
--data_dir /YOUR_PATH/eggnog-mapper-1.0.3/data
# sh hmmer.sh 十几秒就运行完
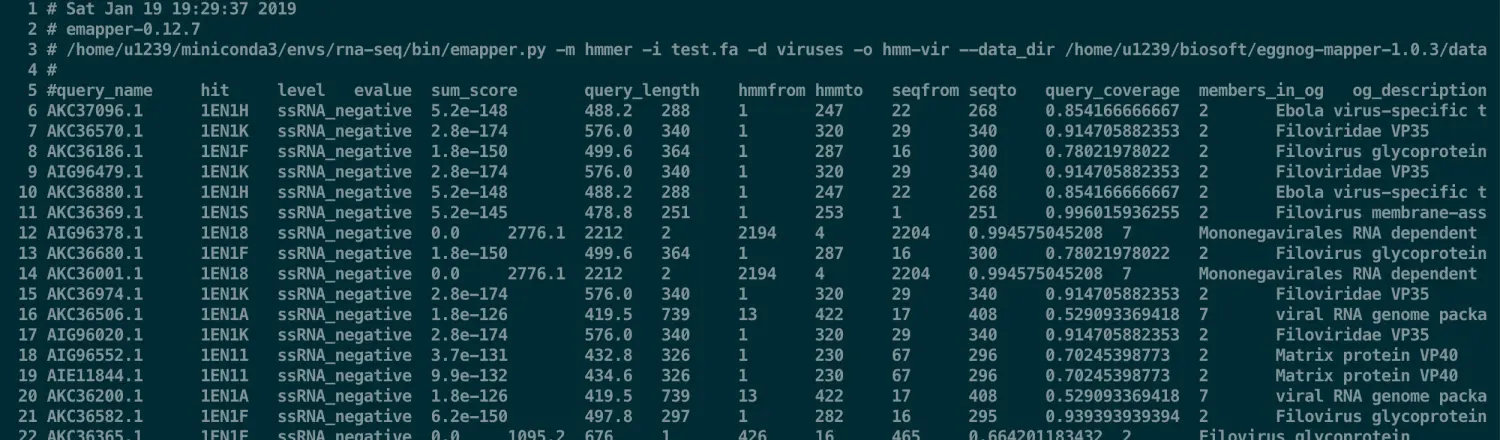
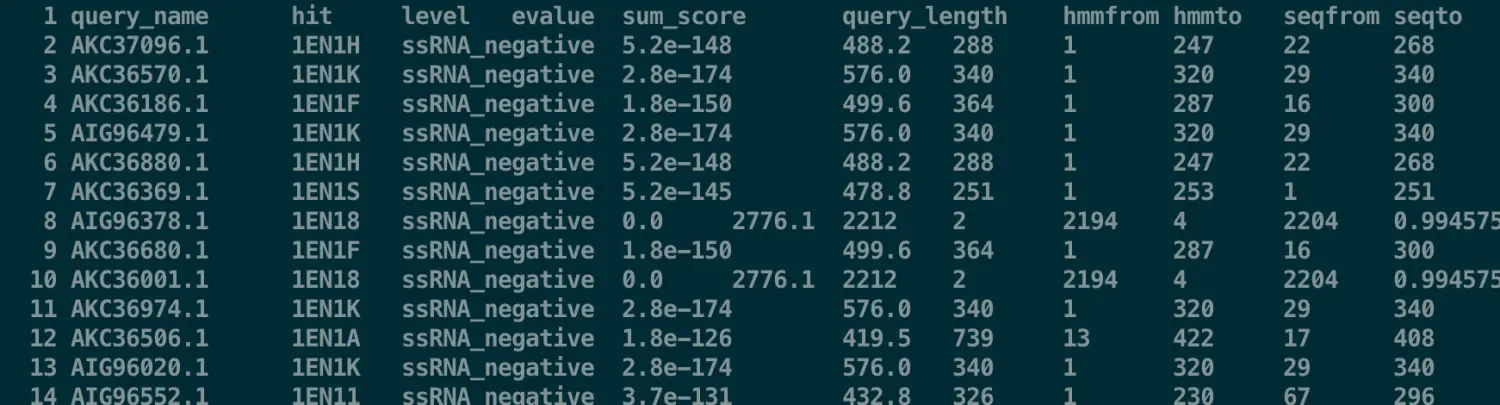
# 然后把结果改一下,去掉#开头的行(但是保留query_name这一行的表头信息)
$ sed -i '/^# /d' hmm-vir.emapper.hmm_hits.annotations
# 然后再去掉query_name前面的#,并且去掉第一行的空行
$ sed -i 's/#//' hmm-vir.emapper.hmm_hits.annotations | sed '/^$/d'
得到了自己的eggnog-mapper结果后,只是搭建了一个桥梁,接下来就是构建自己的OrgDb,再进行GO、KEGG等分析【在OrgDb这一步存在许多选择:首先,如果研究的物种是模式物种,那么直接就可以用bioconductor上的注释包;如果非模式物种而且有人构建过的话,就会上传到AnnotationHub,自己先用
hub <- AnnotationHub()+display(hub)+query(hub, "拉丁名")看看上面有没有;如果也没有,就可以利用eggnog的结果自己进行构建。当然也可以自行构建,然后比较下和别人的结果】
有了OrgDb才能进行富集分析
刘小泽写于19.1.20
要进行GO或者KEGG富集分析,就需要知道每个基因对应什么样的GO/KEGG分类,OrgDb就是存储不同数据库基因ID之间对应关系,以及基因与GO等注释的对应关系的 R 软件包
如果自己研究的物种不在
http://bioconductor.org/packages/release/BiocViews.html#___OrgDb之列,很大可能就需要自己构建OrgDb,然后用clusterProfiler分析所以本次的内容都是基于非模式生物
分类
非模式生物要想找到自己的注释包,又分成两类:
- 一类是在AnnotationHub(https://bioconductor.org/packages/release/bioc/html/AnnotationHub.html)中存在的,例如棉铃虫
- 另一类是在AnnotationHub也不存在相应物种,就需要用AnnotationForge( https://bioconductor.org/packages/release/bioc/html/AnnotationForge.html )来自己构建
第一类:利用AnnotationHub得到org.db
下面我以棉铃虫为例
首先下载并加载AnnotationHub
options("repos"= c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("AnnotationHub", version = "3.8")
library(AnnotationHub)
然后加载现有的物种database
hub <- AnnotationHub() #这一步受限于网速,不成功的话多试几次
# 调用图形界面查看物种
display(hub)
# 或者根据物种拉丁文名称查找
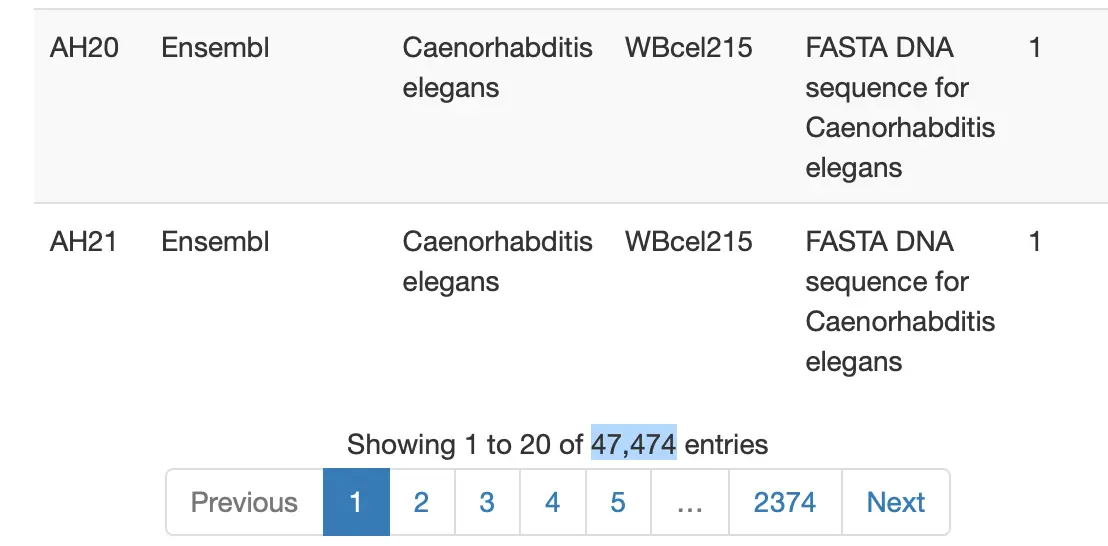
query(hub,"helicoverpa")
# 'object[["AH66950"]]'
title
AH66950 | org.Helicoverpa_armigera.eg.sqlite
AH66951 | org.Heliothis_(Helicoverpa)_armigera.eg....
# 这里AH66950是我们需要的
# 然后下载这个sqlite数据库
ha.db <- hub[['AH66950']]
#查看前几个基因(Entrez命名)
head(keys(ha.db))
#查看包含的基因数
length(keys(ha.db))
#查看包含多少种ID
columns(ha.db)
#查看前几个基因的ID
select(ha.db, keys(ha.db)[1:3],
c("REFSEQ", "SYMBOL"), #想获取的ID
"ENTREZID")
#得到结果
ENTREZID REFSEQ SYMBOL
1 9977712 YP_004021052.1 COX1
2 9977713 YP_004021053.1 COX2
3 9977714 YP_004021054.1 ATP8
#保存到文件
saveDb(ha.db, "Harms-AH66950.sqlite")
#之后再使用直接加载进来
Harms.db <- loadDb("Harms-AH66950.sqlite")
参考:https://mp.weixin.qq.com/s/lHKZtzpN2k9uPN7e6HjH3w
第二类:利用AnnotationForge得到org.db
通过上次的推送( 功能注释整合流程),我们知道了怎样利用eggnog-mapper去人工构建注释、富集桥梁
上次选取的是一个病毒的小例子,这次可以用芝麻(Sesame)做演示
先下载蛋白序列
wget http://www.sesame-bioinfo.org/SesameFG/BLAST_search/G608_contig_2014-08-29.FgeneSH.pep.rar
# 解压后上传
因为统计了下有38406条序列,因此使用diamond来进行功能注释
# 前提是自己安装好eggnog-mapper并且下载好相应的数据库
emapper.py -m diamond \
-i sesame.fa \
-o diamond \
--cpu 19
# 得到如下信息,然后进行处理,只保留表头query_name这一行的注释信息,去掉头尾的# 等信息
sed -i '/^# /d' diamond.emapper.annotations
sed -i 's/#//' diamond.emapper.annotations
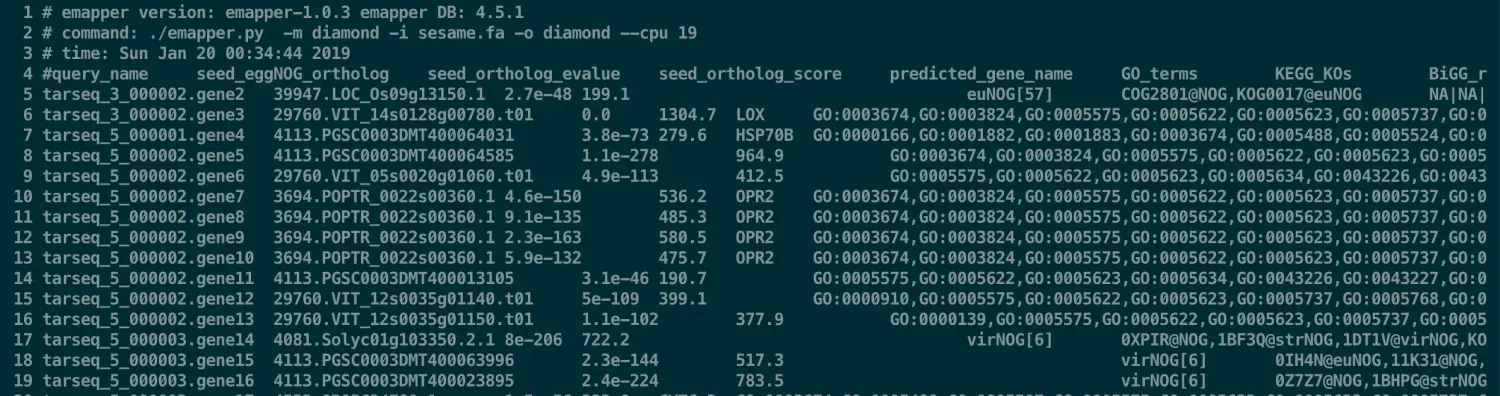
关于结果解释:https://github.com/jhcepas/eggnog-mapper/wiki/Results-Interpretation
其中关于COG的介绍:倒数第二列
COG functional categories: COG functional category inferred from best matching OG 【会给出一个大写字母,每一个大写字母都有自己的解释: COG_explanation】
**STEP1:**自己构建的话,首先需要读入文件
egg_f <- "diamond.emapper.annotations"
egg <- read.csv(egg_f, sep = "\t")
egg[egg==""]<-NA #这个代码来自花花的指导(将空行变成NA,方便下面的去除)
STEP2: 从文件中挑出基因query_name与eggnog注释信息
gene_info <- egg %>%
dplyr::select(GID = query_name, GENENAME = `eggNOG annot`) %>% na.omit()
STEP3-1:挑出query_name与GO注释信息
gterms <- egg %>%
dplyr::select(query_name, GO_terms) %>% na.omit()
**STEP3-2:**我们想得到query_name与GO号的对应信息
# 先构建一个空的数据框(弄好大体的架构,表示其中要有GID =》query_name,GO =》GO号, EVIDENCE =》默认IDA)
# 关于IEA:就是一个标准,除了这个标准以外还有许多。IEA就是表示我们的注释是自动注释,无需人工检查http://wiki.geneontology.org/index.php/Inferred_from_Electronic_Annotation_(IEA)
# 两种情况下需要用IEA:1. manually constructed mappings between external classification systems and GO terms; 2.automatic transfer of annotation to orthologous gene products.
gene2go <- data.frame(GID = character(),
GO = character(),
EVIDENCE = character())
# 然后向其中填充:注意到有的query_name对应多个GO,因此我们以GO号为标准,每一行只能有一个GO号,但query_name和Evidence可以重复
for (row in 1:nrow(gterms)) {
gene_terms <- str_split(gterms[row,"GO_terms"], ",", simplify = FALSE)[[1]]
gene_id <- gterms[row, "query_name"][[1]]
tmp <- data_frame(GID = rep(gene_id, length(gene_terms)),
GO = gene_terms,
EVIDENCE = rep("IEA", length(gene_terms)))
gene2go <- rbind(gene2go, tmp)
}
STEP4-1: 挑出query_name与KEGG注释信息
gene2ko <- egg %>%
dplyr::select(GID = query_name, KO = KEGG_KOs) %>%
na.omit()
STEP4-2: 得到pathway2name, ko2pathway
这一步不需要管代码什么意思,只需要知道它可以帮我们得到以上两个文件就好
if(F){
# 需要下载 json文件(这是是经常更新的)
# https://www.genome.jp/kegg-bin/get_htext?ko00001
# 代码来自:http://www.genek.tv/course/225/task/4861/show
library(jsonlite)
library(purrr)
library(RCurl)
update_kegg <- function(json = "ko00001.json") {
pathway2name <- tibble(Pathway = character(), Name = character())
ko2pathway <- tibble(Ko = character(), Pathway = character())
kegg <- fromJSON(json)
for (a in seq_along(kegg[["children"]][["children"]])) {
A <- kegg[["children"]][["name"]][[a]]
for (b in seq_along(kegg[["children"]][["children"]][[a]][["children"]])) {
B <- kegg[["children"]][["children"]][[a]][["name"]][[b]]
for (c in seq_along(kegg[["children"]][["children"]][[a]][["children"]][[b]][["children"]])) {
pathway_info <- kegg[["children"]][["children"]][[a]][["children"]][[b]][["name"]][[c]]
pathway_id <- str_match(pathway_info, "ko[0-9]{5}")[1]
pathway_name <- str_replace(pathway_info, " \\[PATH:ko[0-9]{5}\\]", "") %>% str_replace("[0-9]{5} ", "")
pathway2name <- rbind(pathway2name, tibble(Pathway = pathway_id, Name = pathway_name))
kos_info <- kegg[["children"]][["children"]][[a]][["children"]][[b]][["children"]][[c]][["name"]]
kos <- str_match(kos_info, "K[0-9]*")[,1]
ko2pathway <- rbind(ko2pathway, tibble(Ko = kos, Pathway = rep(pathway_id, length(kos))))
}
}
}
save(pathway2name, ko2pathway, file = "kegg_info.RData")
}
update_kegg(json = "ko00001.json")
}
STEP5: 利用GO将gene与pathway联系起来,然后挑出query_name与pathway注释信息
load(file = "kegg_info.RData")
gene2pathway <- gene2ko %>% left_join(ko2pathway, by = "KO") %>%
dplyr::select(GID, Pathway) %>%
na.omit()
STEP6: 制作自己的Orgdb
# 查询物种的Taxonomy,例如要查sesame
# https://www.ncbi.nlm.nih.gov/taxonomy/?term=sesame
tax_id = "4182"
genus = "Sesamum"
species = "indicum"
makeOrgPackage(gene_info=gene_info,
go=gene2go,
ko=gene2ko,
pathway=gene2pathway,
version="0.0.1",
outputDir = ".",
tax_id=tax_id,
genus=genus,
species=species,
goTable="go")
sesame.orgdb <- str_c("org.", str_to_upper(str_sub(genus, 1, 1)) , species, ".eg.db", sep = "")
有了Orgdb就可以做富集分析了
# enrichGO最主要的目的就是将基因编号转换成GO号
ego <- enrichGO(gene = id.fc$ENTREZID,
#模式物种
#OrgDb = org.Mm.eg.db,
#非模式物种,例如芝麻
OrgDb = sesame.orgdb,
ont = "BP", #或MF或CC
pAdjustMethod = "BH",
#pvalueCutoff = 0.01,
qvalueCutoff = 0.01)
# 同理也能做enrichKEGG
剩下的可以参考:http://bioconductor.org/packages/release/bioc/vignettes/clusterProfiler/inst/doc/clusterProfiler.html#supported-organisms
最后,如果不想构建orgdb还想做富集分析
满足需求永远是第一生产力,如果你只想用一次,那么clusterProfiler的enricher可以学习一下
主要的两个参数需要注意:gene 是基因ID,TERM2GENE 是GO/KEGG terms与基因ID的对应,例如上面图片中的GO_terms、KEGG_KOs等eggnog-mapper结果,提取出来就好。对于没有对应terms的基因ID,那我们就把它们去掉。
参考:http://guangchuangyu.github.io/2015/05/use-clusterprofiler-as-an-universal-enrichment-analysis-tool/