081-无参转录组知识储备
刘小泽写于19.1.25
最近要开始一批无参转录组学习,我喜欢在学东西时先学一些知识储备,现在不太喜欢拿来代码直接去跑,可能对背后的原理性东西想要做更多了解了。比如这个结果是怎么得到的?怎么看懂结果?我怎么才可以根据自己的需要获得部分结果。这些都需要很长的时间去积累,而流程,只要调试好就可以去跑,但即使不报错的结果也未必是对的,这一点需要注意
先上文章
综述类:
- A survey of best practices for RNA-seq data analysis https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8
- De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3875132/
- RNAseq data analysis from data carpentry
- Great introduction of RNA-seq from sample preparation to data analysis RNA-seqlopedia
- RNA-seq differential expression & pathway analysis with Sailfish, DESeq2, GAGE, and Pathview
细节类:
关于样本数量:How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? https://www.ncbi.nlm.nih.gov/pubmed/27022035
实验设计:https://www.biostars.org/p/65824/
后续标准化、定量、差异分析: Genome-wide repressive capacity of promoter DNA methylation is revealed through epigenomic manipulation
不比对直接定量: sleuth 、 Kallisto 、 Salmon: the sucessor of Salfish ~ Accurate, Versatile and Ultrafast Quantification from RNA-seq Data using Lightweight-Alignment.
转录组分析前期
这部分内容是测序公司经常说的内容,主要考虑以下三点
生物学重复的数量:至少三个以上的生物学重复
文库类型:
- mRNA片段如何获取? 第一种:真核生物成熟的mRNA一般带有polyA尾巴,一般方式就是对具有PolyA尾巴的片段进行捕获。这种方法对mRNA的完整性要求比较高,如果样本发生了降解,那么就会丢失一定的转录组信息 第二种:去除Total RNA中占比很高(超过90%)的rRNA,剩下的RNA中就有mRNA(约1-2%)。样本降解的影响比第一种要小,但需要更高的测序数据量因此成本会高【同时由于原核的mRNA没有polyA,所以只能用去除rRNA的方式】
- 是否需要链特异性文库? 因为现在虽然是叫RNA-seq,但其实还是需要RNA反转录成cDNA然后扩增进行测序,最后是测得还是ATCG。 第一种:利用普通文库。这种方法会同时测到模板链以及反向互补链的信息,不能判断原来的mRNA是什么方向(正链还是负链),既然确定链的来源有偏差,那么也对转录本的定量产生了影响 第二种:利用链特异性文库。这种方法可以将建库过程产生的反向互补序列的文库直接去除掉,可以确定mRNA的来源链,转录本定量的准确性会更高
测序深度:
主要考虑目标转录组的复杂度。低丰度的转录本的定量需要更高的测序深度,但是过高的测序深度也会带来转录本噪声。一般的测序结果中利用饱和曲线可以较好的评估测序深度是否合适
先研究研究流程、文章,最主要用的还是基于比对进行定量的trinity以及不基于比对进行定量的sleuth、kallisto,下一步开始介绍Trinity的使用,这可是一个非常大的套件
刘小泽写于19.2.1
发觉好久没写了,果然临近过年事情比较多,最近重新开始无参转录组。这次先从数据下载做到得到组装的转录本
数据来源
Defining the transcriptomic landscape of Candida glabrata by RNA-Seq.
http://www.ncbi.nlm.nih.gov/pubmed/?term=25586221
这里只下载GSNO和wt的数据SRR1582646-SRR1582651
需要注意的是,如果用自己的数据没有问题,要是使用公共数据的sra再转换,就需要注意一个问题:不要使用平时默认的参数
--gzip --split-3这样会产生这种header:@SRR1582646.224153461,而这种是不被软件识别的我们要使用参数:
--defline-seq '@$sn[_$rn]/$ri' --split-files,这样得到正确的header结果:@HWI-ST1363:114:D1H5CACXX:3:1101:1196:2196/1
数据下载转换
SRR_Acc_List.txt中放入SRR的ID号
SRR1582646 SRR1582647 SRR1582648 SRR1582649 SRR1582650 SRR1582651
wkd=~/rna-denovo-test
source activate rna-seq
mkdir $wkd/raw && cd $wkd/raw
cat $wkd/raw/SRR_Acc_List.txt |while read i
do
ascp -v -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh -k 1 -T \
-l200m anonftp@ftp-private.ncbi.nlm.nih.gov:/sra/sra-instant/reads/ByRun/sra/SRR/SRR158/$i/${i}.sra ./
time fastq-dump --defline-seq '@$sn[_$rn]/$ri' --split-files -A $i ${i}.sra && echo "** ${i}.sra to fastq done **"
done
source deactivate
预处理(质控+过滤)
因为是测试数据,因此我们可以只选择其中10k行,但是为了选取的随机性,可以使用seqtk软件
source activate rna-seq
# 选择一部分10000条,用seqtk sample随机选10000条
ls $wkd/raw/*_1.fastq.gz >1
ls $wkd/raw/*_2.fastq.gz >2
paste 1 2 > sample-conf
cat conf1 | while read i;do
fqs=($i)
fq1=${fqs[1]}
fq2=${fqs[2]}
surname=${fq1%%_*}
#echo $fq1 $surname
seqtk sample $fq1 10000 >test.${surname}_1.fastq
seqtk sample $fq2 10000 >test.${surname}_2.fastq
done
第一次质控
mkdir $wkd/qc && cd $wkd/qc
fastqc -o ./ -t 10 $wkd/raw/*.fastq
multiqc ./
source deactivate
过滤
source activate rna-seq
mkdir $wkd/clean && cd $wkd/clean
ls $wkd/raw/*_1.fastq >1
ls $wkd/raw/*_2.fastq >2
paste 1 2 > filter_conf
cat filter_conf | while read i;do
fqs=($i)
fq1=${fqs[0]}
fq2=${fqs[1]}
trim_galore -q 25 --phred33 --length 50 -e 0.1 --stringency 3 \
--paired -o $wkd/clean $fq1 $fq2
done
source deactivate
再一次质控
cd $wkd/clean
source activate rna-seq
fastqc -o ./ -t 10 $wkd/clean/*.fq
multiqc ./
source deactivate
Trinity拼接
如果分析的数据多,就可以做个表格;数据少的话,直接指定–left,–right
这里在正式分析前,先做一个样本表格samples.txt,其中包含了处理、重复、左端数据、右端数据
# cond_A cond_A_rep1 A_rep1_left.fq A_rep1_right.fq
# cond_A cond_A_rep2 A_rep2_left.fq A_rep2_right.fq
# cond_B cond_B_rep1 B_rep1_left.fq B_rep1_right.fq
# cond_B cond_B_rep2 B_rep2_left.fq B_rep2_right.fq
mkdir $wkd/assembly && cd $wkd/assembly
echo -e "GSNO\nGSNO\nGSNO\nwt\nwt\nwt" >1
echo -e "GSNO_SRR1582646\nGSNO_SRR1582647\nGSNO_SRR1582648\nwt_SRR1582649\nwt_SRR1582650\nwt_SRR1582651" >2
ls $wkd/clean/*1_val_1.fq >3
ls $wkd/clean/*2_val_2.fq >4
paste 1 2 3 4 >samples.txt
跑程序很简单,需要指定样本文件、CPU、内存、min_contig_length(minimum assembled contig length)【指定拼接最小长度,默认200bp,低于这个长度的contig就被丢弃】
Trinity --seqType fq --samples_file $wkd/assembly/samples.txt \
--CPU 10 --max_memory 10G --min_contig_length 150
运行Trinity中间会用到samtools,但是有时候安装它会失败,会遇到动态库的问题(就是某些lib缺失)。最简单的办法就是降级,如最新版1.9,我们安装1.8 ,例如
conda install samtools=1.8
剩下的运行过程中 ~1 hour and ~1G RAM per ~1 million PE reads
首先运行第一个程序:seqtk-trinity ,目的是将fq转为fa,并生成both.fa
第二个程序:Jellyfish ,中间过程jellyfish count + histo + dump,starting with Jellyfish to generate the k-mer catalog
第三个程序:Inchworm, assemble ‘draft’ contigs
第四个程序:Chrysalis,cluster the contigs and build de Bruijn graphs
第五个程序:Butterfly,tracing paths through the graphs and reconstructing the final isoform sequences
最后的结果就是得到了Trinity.fasta,如
>TRINITY_DN506_c0_g1_i1 len=171 path=[149:0-170] [-1, 149, -2]
TGAGTATGGTTTTGCCGGTTTGGCTGTTGGTGCAGCTTTGAAGGGCCTAAAGCCAATTGT
TGAATTCATGTCATTCAACTTCTCCATGCAAGCCATTGACCATGTCGTTAACTCGGCAGC
AAAGACACATTATATGTCTGGTGGTACCCAAAAATGTCAAATCGTGTTCAG
>TRINITY_DN512_c0_g1_i1 len=168 path=[291:0-167] [-1, 291, -2]
ATATCAGCATTAGACAAAAGATTGTAAAGGATGGCATTAGGTGGTCGAAGTTTCAGGTCT
AAGAAACAGCAACTAGCATATGACAGGAGTTTTGCAGGCCGGTATCAGAAATTGCTGAGT
AAGAACCCATTCATATTCTTTGGACTCCCGTTTTGTGGAATGGTGGTG
其中header信息中包含了:转录本名称 、长度、 de Bruijn图重构路径,其中g表示gene,i表示isoform转录本,多个转录本可以重构成一个基因,在下游差异分析中,可以基于基因或转录本两种层次进行分析
刘小泽写于19.2.2
今天主要看看转录本拼接质量评估
前言
我们利用trinity简单的命令拼接好转录本后,想要知道我们拼接的质量如何,如果不好可能需要寻找原因,重新调整参数再重新拼接。这一步是很关键的,因为拼接的转录本相当于有参里面的参考转录组,试想如果参考都做不好,那岂不是上梁不正下梁歪?
首先我们可以看看组装了多少条转录本
grep '>' $wkd/assembly/trinity_out_dir/Trinity.fasta | wc -l
但这仅仅是最粗略的方式,因为随着测序深度的加大,得到的contigs越多,因此可以拼接更多的转录本
另外可以看看基本的统计值
TrinityStats.pl $wkd/assembly/trinity_out_dir/Trinity.fasta
第一部分Counts of transcripts, etc. 结果中的 Total trinity transcripts 和上面那个命令结果一致【正常数据中转录本数量小于20w是正常的,如果数量达到了30w、40w条,就需要先用corset软件进行聚类】
结果中还有组装的gene数 Total trinity ‘genes’ ,可以看到transcripts的数量比genes的数量多,因为存在一个基因的可变剪切(这种情况在昆虫和哺乳动物中比较常见)
另外还有第二部分内容Stats based on ALL transcript contigs: N50值(at least half of the assembled bases are in contigs of at least that contig length
累加后长度超过转录组总长度一半的contig的长度就是N50)
【正常情况下,N50应该是1k左右】
说到评估,我们一般会想到:和其他拼接工具的结果进行对比(就好像在有参中使用3-5中工具进行比对,看看分别的比对率),或者使用不同的参数看看结果异同(前提是对参数的设置比较了解) 总的来说,上面的方法得到的结果不是特别有用,有下面几种方法可以更有效地评价转录本质量
第一种 align
将reads重新比对到转录本上,看看比对率
一般来讲,至少应该有80%的原始数据可以在拼接的转录本中找到(经验值),剩下的没有拼接上的序列可能由于低表达导致没有足够的覆盖度进行拼接或者序列质量较低或者重复reads。
比对过程中,除了正确比对PE reads,还有可能仅仅一条read比对上,或者由于序列太短比对不上,如果利用bowtie、STAR进行比对,仅仅会计算成功比对上的PE reads。这里为了客观评价,我们需要知道所有的比对情况(包括单端比对和未比对上),可以利用Bowtie2
首先为转录本构建索引
bowtie2-build Trinity.fasta Trinity.fasta
然后进行双端比对,输出的sam转为bam
bowtie2 -p 10 -q --no-unal -k 20 -x Trinity.fasta -1 reads_1.fq -2 reads_2.fq \
2>align_stats.txt| samtools view -@10 -Sb -o bowtie2.bam
#-p 线程数
#-q input文件为fq(默认)
#--no-unal输出的sam文件中不记录未必对的reads信息
#-k 指定每条read匹配上的前20个记录,按匹配分值降序排列(descending order by alignment score)this mode can be effective and fast when user cares more about whether a read aligns (or aligns a certain number of times) than where exactly it originated.
bowtie2说明书:http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml
可以将比对结果放入IGV中查看,但首先需要排序、构建索引
ls $wkd/evaluate/1-bowtie2/*.bam | while read i; do
file=`basename $i`
#echo $file
surname=${file%%.bowtie2.bam}
#echo $surname
samtools sort $i -o $wkd/evaluate/1-bowtie2/${surname}.sorted.bam
samtools index $wkd/evaluate/1-bowtie2/${surname}.sorted.bam
done
samtools faidx $wkd/assembly/trinity_out_dir/Trinity.fasta
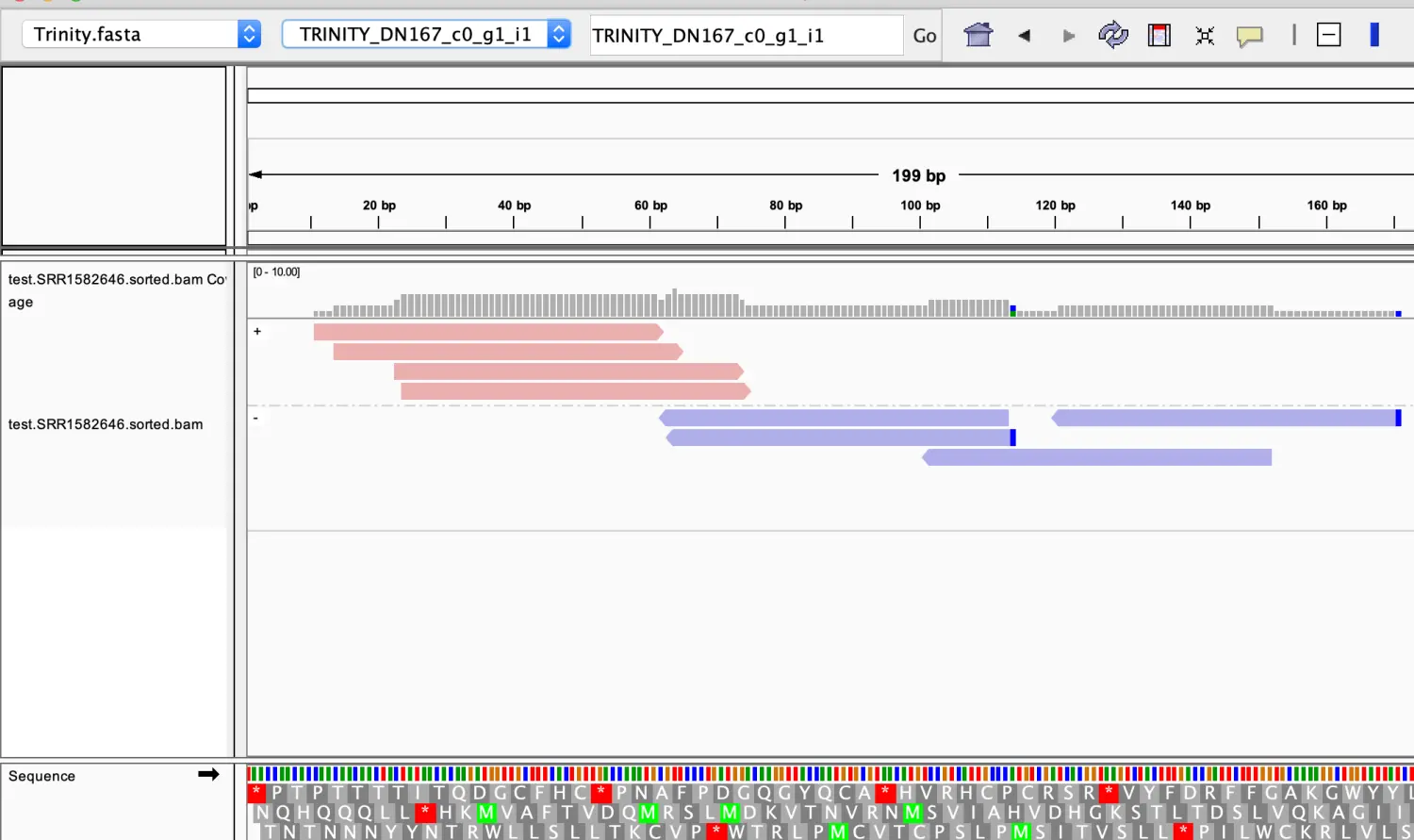
上面因为是选取的10000条序列,因此比对的结果一般,实际的数据更应该接近于下图
第二种 uniprot
将拼接转录本与已知蛋白序列库进行比对
核心思想就是检查拼接好的全长或者接近全长的转录本序列数目(因为这样的序列才有比对结果)。这种方法如果用在有参考转录本的模式物种(如人、小鼠)是很简单的,直接比对到参考转录组然后检查比对上的序列长度区间;非模式物种可以比对到近源物种的参考转录组上进行查看;更为一般的做法,就是将拼接的转录本比对到已知的蛋白数据库,看看能比对到多少蛋白序列,从而推断拼接质量
利用blast+和蛋白数据库 SwissProt、 TrEMBL 【关于这两个数据库:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC102476/】
简而言之, SWISS-PROT: curated, high level of annotation, minimal level of redundancy ; TrEMBL: computer-annotated upplement to SWISS-PROT, derived from the translation of all coding sequences (CDSs) in the EMBL except the CDSs already included in SWISS-PROT
#Build a blastable database:
makeblastdb -in uniprot_sprot.fasta -dbtype prot
# Perform the blast search, reporting only the top alignment:
cp $wkd/assembly/trinity_out_dir/Trinity.fasta ./
blastx -query Trinity.fasta -db uniprot_sprot.fasta -out blastx.outfmt6 \
-evalue 1e-20 -num_threads 6 -outfmt 6
# -max_target_seqs 1 这个参数可以加上试试 Maximum number of aligned sequences to keep
关于blast参数(- max_target_seqs)的设置还专门有一篇文章讲重要性:https://academic.oup.com/bioinformatics/advance-article-abstract/doi/10.1093/bioinformatics/bty833/5106166 结论就是:这个max_target_seqs参数有bug,不懂就用默认
检查结果
distribution of percent length coverage for the top matching database entries就是比对蛋白结果的序列长度分布,这么说可能有点拗口,简单理解就是:最左侧一列是蛋白序列比对到Trinity拼接转录本的覆盖度,第2列是落在某个覆盖度范围内的蛋白序列个数,第三列是一个蛋白数目累加值
analyze_blastPlus_topHit_coverage.pl blastx.outfmt6 Trinity.fasta uniprot_sprot.fasta
# 参数设置: blast+.outfmt6.txt query.fasta search_db.fasta
cat blastx.outfmt6.hist
#hit_pct_cov_bin count_in_bin >bin_below
100 71 71
90 22 93
80 11 104
70 11 115
60 19 134
50 22 156
40 38 194
30 25 219
20 62 281
10 14 295
如果比对的目标蛋白序列同时匹配多个Trinity转录本序列,并且都是best hits,那么结果也只计作一次作为highest BLAST bit score和longest match length
(Only the single best matching Trinity transcript is reported for each top matching database entry)
上面的结果主要理解成:
- 有22个蛋白,它们的序列的**80%~90%**可以比对到Trinity拼接的转录本
- 有93个蛋白,它们有超过80%的序列覆盖度(即可以比对到Trinity拼接的转录本),可以认为有93个比对到了全长转录本
- 有71个蛋白,它们超过**90%**的序列覆盖度
同样,可以进行Nr, Pfam,Nt数据库的比对,只要下载数据库=》建库=》比对,当然,如果下载的数据库(如:NCBI的nt、nr)已经构建好了索引,就可以跳过建库的步骤
统计升级
blast的过程存在这种情况:一条转录本比对到一个蛋白序列,另外还有几个不连续的片段(即:multiple high scoring segment pairs (HSPs))。上面的blast流程只考虑了single best alignment这种情况,我们可以将每个转录本的HSPs组合在一起,然后基于此进行覆盖度计算
# Group the multiple HSPs
$TRINITY_HOME/util/misc/blast_outfmt6_group_segments.pl blastx.outfmt6 \
Trinity.fasta uniprot_sprot.fasta > blastx.outfmt6.grouped
# compute the percent coverage by length histogram
blast_outfmt6_group_segments.tophit_coverage.pl blastx.outfmt6.grouped
#hit_pct_cov_bin count_in_bin >bin_below
100 72 72
90 22 94
80 11 105
70 11 116
60 19 135
50 23 158
40 37 195
30 24 219
20 61 280
10 14 294
虽然得到的结果和上面单独blast差不多,但是目前是基于grouped HSPs/pair而非single HSP/pair
第三种 BUSCO
在近缘物种中,总有一些基因是保守的,BUSCO 软件根据OrthoDB 数据库,构建了几个大的进化分支的单拷贝基因集。将转录本拼接结果与该基因集进行比较,根据比对上的比例、完整性,来评价拼接结果的准确性和完整性。
官网:https://busco.ezlab.org/
安装:conda install busco
目前支持的数据库列表如下,选择研究物种的数据库进行下载。不同的数据库,评价结果可能完全不同。

mkdir $wkd/evaluate/5-busco && cd $wkd/evaluate/5-busco
# 下载数据库
wget http://busco.ezlab.org/v2/datasets/fungi_odb9.tar.gz
# 提取每个基因的最长转录本
get_longest_isoform_seq_per_trinity_gene.pl $wkd/assembly/trinity_out_dir/Trinity.fasta \
>longest_isoform.fasta
# 运行 BUSCO
# -l/--lineage: 数据库的路径
# -m:运行模式,geno|tran|prot
busco -i longest_isoform.fasta \
-l euarchontoglires_odb9 \
-o busco \
-m tran \
--cpu 2
结果主要看统计文件run_busco/short_summary_busco.txt,这里尽量选用真实数据进行测试,数据量太少得到的结果不准确
结果类似于:
# Summarized benchmarking in BUSCO notation for file longest_isoform.fasta
# BUSCO was run in mode: tran
C:90.0%[S:47.9%,D:38.2%],F:3.4%,M:6.7%,n:439
383 Complete BUSCOs (C)
216 Complete and single-copy BUSCOs (S)
167 Complete and duplicated BUSCOs (D)
20 Fragmented BUSCOs (F)
27 Missing BUSCOs (M)
430 Total BUSCO groups searched
C值(Complete BUSCOs)表示和BUSCO集相比的完整度,一般能达到80%以上。但是如果达不到也不需要担心,可以再多组装几个版本,选个最优解
关于转录本定量
一般有两种方式,一个是基于比对定量(如RSEM、eXpress);另一种是不基于比对的方法(转录本划分成kmer, 用kmer出现的次数来衡量转录本丰度,如kallisto、salmon,优点就是快)
注意一:如果自己有多组数据(例如同一物种的不同组织),最好先全部拼接成一个转录本,然后再分开进行定量
注意二:Trinity软件虽然可以调用RSEM、eXpress等,但是并不是内置,还是需要我们自己先手动安装好,放入环境变量;或者直接conda安装
Trinity为了定量,推出了一个脚本align_and_estimate_abundance.pl
帮助文档很详细,需要注意的就是: 尽量使用--samples_file这个参数,因为它会将不同的处理及重复的结果文件自动放入不同的目录;--prep_reference这个参数是很有帮助的,它先对拼接的转录本构建索引,然后再对不同样本进行并行计算表达量,可以视作一个提速的手段;--trinity_mode或者--gene_trans_map 除了得到isoform的表达矩阵,还可以得到gene层面的表达矩阵【前面提到过,拼接的转录本中一个gene对应多个isoform】
怎么定量?
基于比对的模式生成的结果中会有FPKM(fragments per kilobase transcript length per million fragments mapped)、TPM(transcripts per million transcripts)两种标准化方法;不基于比对的结果只有TPM。
所谓的标准化就是:去除测序深度和基因长度的的影响
这两种哪种更合理呢?关于FPKM和TPM之争,许多文章都有介绍: Wagner et al. 2012. Theory Biosci , Li and Dewey, BMC Bioinf. 2011. 目前一般都推荐TPM
首先,回归主要问题:定量。我们来看看什么是表达量?
- 如果要明确知道一个细胞中、或者是一定摩尔量的 RNA中,有多少条某种转录本,就是绝对定量(如果能够对样品进行细胞计数或者添加 spike-in, 转录组也可以实现绝对定量)
- 但我们平时做的转录组测序,要求并没那么高,并不知道用于建库测序的 RNA 来自多少个细胞,总共有多少转录本。因此只能相对定量,也就是描述某基因的转录本占样本中所有转录本的百分比。
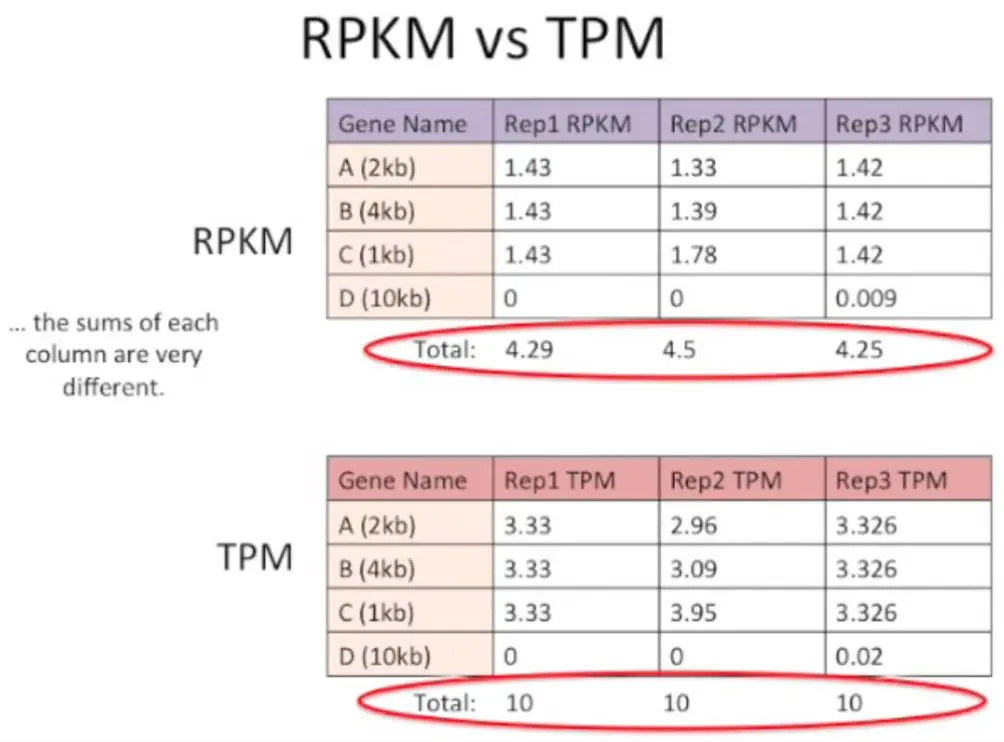
好了,前面说的定量就是统计的read count数(下图左一),但是这还并不能说明真实情况,因为没有考虑长度的分布(比如下图的3和4:在read count总数中确实4比3要多许多,但是注意观察,3是两段短的转录本,4是一个长一短。有没有注意到,其实3和4的read分布是一个水平的,它们都很均匀,和1相比1就显得比较稀疏)。为了更好了了解表达量(Expression)和片段数(read count)的差别,我们再来看看下面图中的2和4,总量的话(左一)2和4是一样的,但是2明显每个转录本分布的reads更多,因此换算到表达量时,2的表达量要比4高不少(右一)
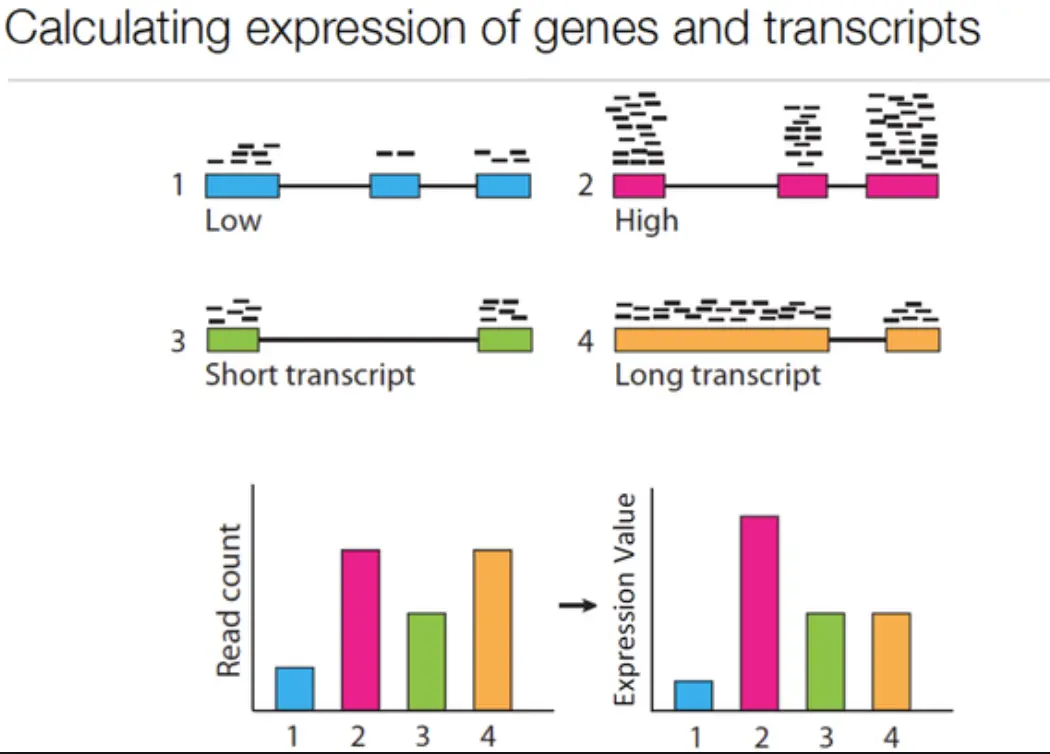
上面知道了要同时考虑raw count和转录本长度的影响,因此就需要公式来进行标准化,公式很多,那么哪一种更能反映真实问题呢?

对公式不清楚的话,其实简单看发表年份就能知道,TPM比FPKM更先进一些
下面具体从公式角度了解,看看两种方法的计算公式:
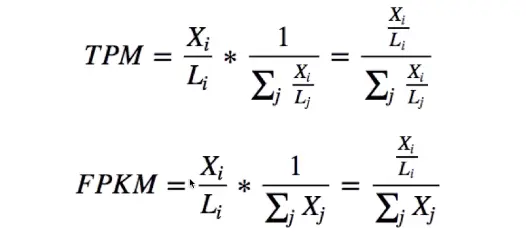
Xi表示比对到某基因上的Fragment数目,单位是Millions Fragments;Li表示这个基因外显子的长度,单位是Kilobase。因此Xi/Li 的商就是外显子长度为1Kb的基因Fragment数目,即转录本丰度,因此TPM可以校正转录本长度分布的影响,用于可以更好地进行样本间比较。
【注意,二者分母不同,TPM的分母中考虑了长度的影响,可以理解成总的转录本丰度;而FPKM中,没有考虑长度影响,可以理解成总的Fragment数目】
如果对每个样品总 表达量求和,会发现 TPM中各个样本的总和是相等的,而FPKM则不相等。因此,能用TPM就不用FPKM
【目前DESeq2和edgeR不用RPKM/FPKM或TPM做均一化,而是直接用原始的read counts做均一化处理】
参考:https://haroldpimentel.wordpress.com/2014/05/08/what-the-fpkm-a-review-rna-seq-expression-units/
https://www.youtube.com/watch?v=TTUrtCY2k-w
定量结果都有啥?
基于比对的定量
比对过程会产生bowtie.bam,这个文件会直接导入RSEM或eXpress,如果定量时选择--coordsort_bam参数,会对bam文件进行sort,生成一个bowtie.csorted.bam文件,为了方便IGV可视化
RSEM(RNA-Seq by Expectation Maximization):结果生成两个文件,均包含定量信息:
结果产生两个文件 RSEM.isoforms.results : 每个拼接转录本的最大期望count数[见下表] RSEM.genes.results : 一个基因对应多个转录本(此处基因非严格意义上的基因),统计了每个"基因“的最大期望count数transcript_id gene_id length effective_length expected_count TPM FPKM IsoPct TRINITY_DN100_c0_g1_i1 TRINITY_DN100_c0_g1 443 181.37 4.84 1670.06 12311.85 100.00 TRINITY_DN101_c0_g1_i1 TRINITY_DN101_c0_g1 251 19.37 1.00 3231.22 23820.87 100.00 TRINITY_DN103_c0_g1_i1 TRINITY_DN103_c0_g1 1219 957.37 0.00 0.00 0.00 100.00 TRINITY_DN103_c0_g2_i1 TRINITY_DN103_c0_g2 414 152.41 0.00 0.00 0.00 100.00 关于RSEM:https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-12-323
eXpress:
结果产生两个文件(10 tab-delimited columns, sorted by the bundle_id) results.xprs : transcript abundance estimates (generated by eXpress) results.xprs.genes : gene abundance estimates (provided here by summing up transcript values per gene)包含了: estimated counts, FPKM, and TPM 等信息
关于eXpress:https://pachterlab.github.io/eXpress/overview.html#top
帮助文档:https://pachterlab.github.io/eXpress/manual.html
不基于比对的定量
Kallisto:
结果产生两个文件 abundance.tsv : transcript abundance estimates (generated by kallisto) abundance.tsv.genes : gene abundance estimates (provided here by summing up transcript values per gene)输出结果短小精悍
target_id length eff_length est_counts tpm TRINITY_DN10_c0_g1_i1 334 100.489 13 4186.62 TRINITY_DN11_c0_g1_i1 319 87.9968 0 0 TRINITY_DN12_c0_g1_i1 244 38.2208 2 1693.43 TRINITY_DN17_c0_g1_i1 229 30.2382 5 5351.21 Salmon:
结果产生两个文件 quant.sf : transcript abundance estimates (generated by salmon) quant.sf.genes : gene-level abundance estimates (generated here by summing transcript values)结果也是很简单
Name Length EffectiveLength TPM NumReads TRINITY_DN10_c0_g1_i1 334 135.084 0 0 TRINITY_DN11_c0_g1_i1 319 120.926 6158.51 13 TRINITY_DN12_c0_g1_i1 244 57.9931 0 0 TRINITY_DN17_c0_g1_i1 229 47.8216 2395.84 2
组合定量结果
因为之前是每个sample生成一个结果文件,现在需要组合在一起,使用abundance_estimates_to_matrix.pl
需要指定的参数:
--est_method <string> RSEM|eXpress|kallisto|salmon (needs to know what format to expect)需要和之前定量方法一致才能识别并组合
--gene_trans_map <string> the gene-to-transcript mapping file. (if you don't want gene estimates, indicate 'none').
例如:
ls */RSEM.genes.results >genes.quant_files.txt
$TRINITY_HOME/util/abundance_estimates_to_matrix.pl
--est_method RSEM \
--cross_sample_norm TMM \
--out_prefix rsem-gene \
--name_sample_by_basedir \
--quant_files genes.quant_files.txt
得到结果
rsem-gene.isoform.counts.matrix : the estimated RNA-Seq fragment counts (raw counts)
rsem-gene.isoform.TPM.not_cross_norm : a matrix of TPM expression values (not cross-sample normalized)
rsem-gene.isoform.TMM.EXPR.matrix : a matrix of TMM-normalized expression values
其中counts.matrix文件是用来下游差异表达分析的 , TMM.EXPR.matrix 文件是基因表达矩阵
关于TMM的重要性: Robinson & Oshlack, Genome Biology 2010 and Dillies et al., Brief Bioinf, 2012
计算表达的转录本或基因数量
使用PtR作图
使用Trinity组装转录本,并定量后,有了基因或者转录本的表达量矩阵,先不用着急去做差异分析,可以先检查下数据,例如:组内生物学重复间相关性是不是高于组间重复,计算基因间的相关性,样本热图、基因热图、PCA分析
其实有了表达矩阵,自己用代码做也是可以的,PtR这个程序就是使用更方便而已
生物学重复之间比较
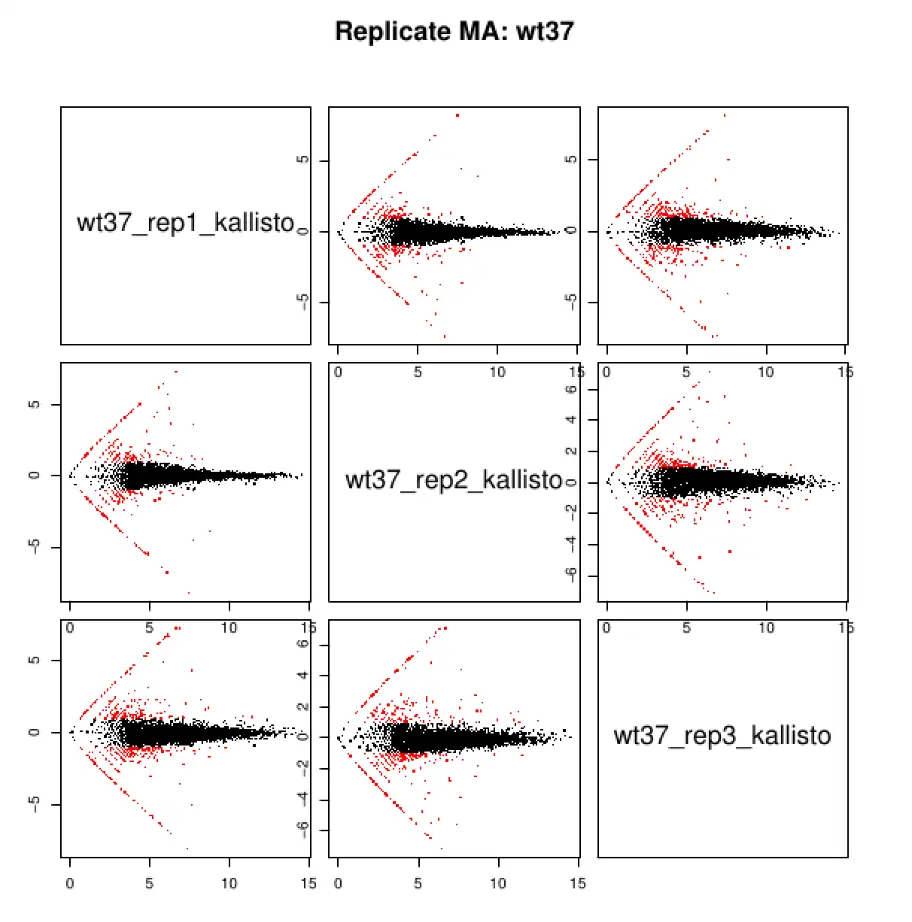
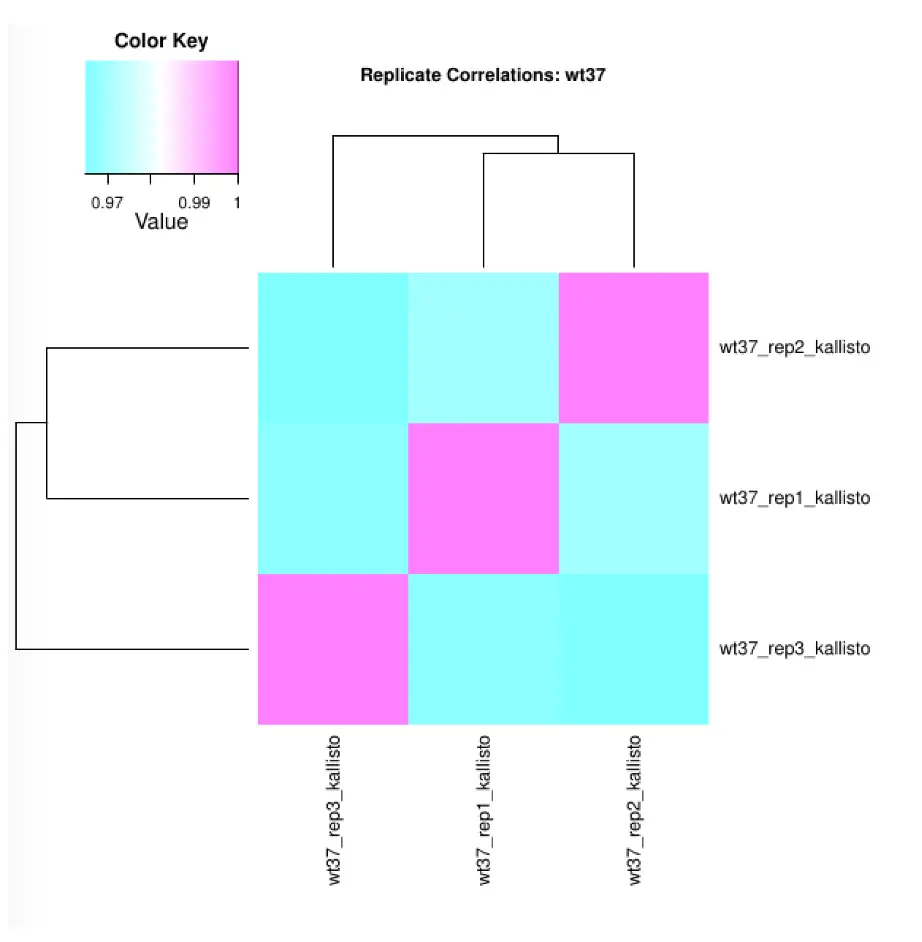
$TRINITY_HOME/Analysis/DifferentialExpression/PtR \ -m Trinity_trans.counts.matrix -s samples.txt \ --log2 --compare_replicates结果每个组生成一个pdf文件,每个pdf文件中有四张图片
第一张:Counts of reads mapping to transcripts

第二张:Pairwise scatter plot of log2(read counts) per transcript

第三张:Pairwise MA plots
第四张:Pearson correlation between log2(replicate counts)
比较所有样本中各个重复的相关性,就是进行聚类分析
$TRINITY_HOME/Analysis/DifferentialExpression/PtR \ -m Trinity_trans.counts.matrix -s samples.txt \ --log2 --sample_cor_matrix结果生成:Trinity_trans.counts.matrix.log2.sample_cor_matrix.pdf
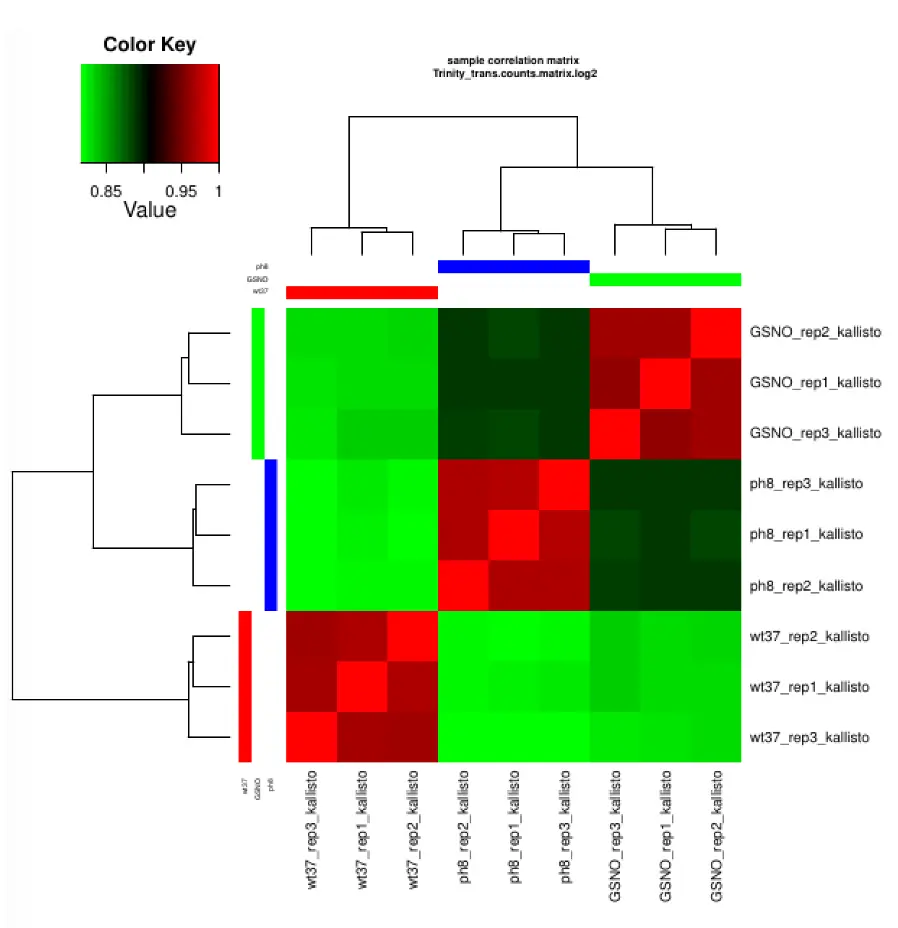
PCA
$TRINITY_HOME/Analysis/DifferentialExpression/PtR\ -m Trinity_trans.counts.matrix -s samples.txt\ --log2 --CPM --prin_comp 3
着重看下重复是不是按照样本聚在一起,如果看到按照测序时间或其他因素聚在一起,那么就可能存在批次效应,应该进行去除
寻找可能的生物学功能信息
刘小泽写于19.2.17
得到了组装的转录本,就可以根据这个去找潜在的生物学相关信息,比如编码区预测、注释等
利用 TransDecoder 这个工具可以在转录本序列中寻找潜在的编码区,它先找到长的开放阅读框区域(ORFs)
TransDecoder.LongOrfs -t Trinity.fasta
然后根据序列组成打分
TransDecoder.Predict -t Trinity.fasta
结果会得到一些带有transdecoder字符的文件
Trinity.fasta.transdecoder.bed
Trinity.fasta.transdecoder.cds
Trinity.fasta.transdecoder.gff3
Trinity.fasta.transdecoder.pep
Trinity.fasta.transdecoder_dir/
...
其中最关心的就是:Trinity.fasta.transdecoder.pep ,其中包含了预测的编码区对应的蛋白序列
$ less Trinity.fasta.transdecoder.pep
>TRINITY_DN107_c0_g1_i1.p1 TRINITY_DN107_c0_g1~~TRINITY_DN107_c0_g1_i1.p1 ORF type:internal len:175 (+),score=164.12 TRINITY_DN107_c0_g1_i1:2-523(+)
VPLYQHLADLSDSKTSPFVLPVPFLNVLNGGSHAGGALALQEFMIAPTGAKSFREAMRIG
SEVYHNLKSLTKKRYGSSAGNVGDEGGVAPDIQTAEEALDLIVDAIKAAGHEGKVKIGLD
CASSEFFKDGKYDLDFKNPNSDASKWLSGPQLADLYHSLVKKYPIVSIEDPFAE
>TRINITY_DN10_c0_g1_i1.p2 TRINITY_DN10_c0_g1~~TRINITY_DN10_c0_g1_i1.p2 ORF type:internal len:158 (-),score=122.60 TRINITY_DN10_c0_g1_i1:2-472(-)
TDQDKRYQAKMGKSHGYRSRTRYMFQRDFRKHGAIALSTYLKVYKVGDIVDIKANGSIQK
GMPHKFYQGKTGVVYNVTKSSVGVIVNKMVGNRYLEKRLNLRVEHVKHSKCRQEFLDRVK
SNAAKRAEAKAQGKAVQLKRQPAQPREARVVSTEGNV
这个文件中:header行包含了蛋白的ID信息、原始转录本ID信息、type信息、长度、正负链、打分信息、ORF坐标信息
其中type信息可能会出现:
- complete:包含起始、终止密码子
- 5prime_partial:丢失起始密码子,可能是N端的一部分
- 3prime_partial:丢失终止密码子,可能是C端的一部分
- internal:既有N端又有C端的部分
得到的.pep文件可以用于接下来的同源比对
刚得到转录本时就进行了一次SWISSPROT数据库的比对,目的是找到全长转录本;这里再利用得到的蛋白序列进行同源比对,只不过需要适当降低evalue,因此目前序列较之前转录本少许多
blastp -query Trinity.fasta.transdecoder.pep \
-db mini_sprot.pep -num_threads 5 \
-max_target_seqs 1 -outfmt 6 -evalue 1e-5 \
> swissprot.blastp.outfmt6
另外可以用HMMER进行一个Pfam比对,找到可能的保守结构域
hmmscan --cpu 5 --domtblout TrinotatePFAM.out \
Pfam-A.hmm \
Trinity.fasta.transdecoder.pep
可以预测下信号肽、跨膜结构域信息
使用signalp预测信号肽
signalp -f short -n signalp.out Trinity.fasta.transdecoder.pep
看下结果
$ less signalp.out
##gff-version 2
##sequence-name source feature start end score N/A ?
## -----------------------------------------------------------
TRINITY_DN19_c0_g1_i1|m.141 SignalP-4.0 SIGNAL 1 18 0.553 . . YES
TRINITY_DN33_c0_g1_i1|m.174 SignalP-4.0 SIGNAL 1 19 0.631 . . YES
使用wc -l 可以看预测了多少的信号肽
还可以使用tmhmm 预测跨膜结构区域
关于各种预测的软件信息:http://www.cbs.dtu.dk/services/software.php
转录本的统计指标N50 vs ExN50
刘小泽写于19.2.19
之前得到转录本的时候,我们使用TrinityStats.pl Trinity.fasta这个命令对转录本进行了统计,结果包含了Contig Nx长度的统计,例如:
$TRINITY_HOME/util/TrinityStats.pl Trinity.fasta
################################
## Counts of transcripts, etc.
################################
Total trinity 'genes': 1388798
Total trinity transcripts: 1554055
Percent GC: 44.52
########################################
Stats based on ALL transcript contigs:
########################################
Contig N10: 5264
Contig N20: 3136
Contig N30: 1803
Contig N40: 989
Contig N50: 606
Median contig length: 288
Average contig: 511.61
Total assembled bases: 795066996
Contig N10: 5264表示至少10%的转录本序列拥有至少5264个碱基长度。这个结果文件中我们一般关注N50这个指标,也就是说组装结果至少一半的转录本序列的长度。
当然,基于全部contigs的统计方式有一个问题:如果转录本比较长,并且拼接得到的isoform太多,那么N50会变大【因为Nx的计算方式是:先将长度从大到小排列,然后计算总长度,依次累加contig,看哪个contig先达到总长一半,就作为N50】,但这并不能真实反映拼接情况,因此还有一个选择:每个基因选择最长的一条isoform进行统计
#####################################################
## Stats based on ONLY LONGEST ISOFORM per 'GENE':
#####################################################
Contig N10: 3685
Contig N20: 1718
Contig N30: 909
Contig N40: 588
Contig N50: 439
Median contig length: 281
Average contig: 433.39
Total assembled bases: 601896081
可以看到,这种方式比一开始得到的N50要小,正是降低了”一条基因有多条长的isoform“这种影响,因此如果使用N50的话,推荐使用ONLY LONGEST ISOFORM per 'GENE'这个指标中的
相比N50,还有比它更合适的ExN50 ,多了个Ex,意思就是Expressed,要看表达量
ExN50除了考虑上面N50的计算方法,还限制了必须是最高表达的转录本(代表了前x%的总表达量)
$TRINITY_HOME/util/misc/contig_ExN50_statistic.pl \
transcripts.TMM.EXPR.matrix Trinity.fasta >ExN50.stats
结果会生成这样的一个表格,第一列是Ex表达量(%),第二列是ExN50,第三列是num_transcripts
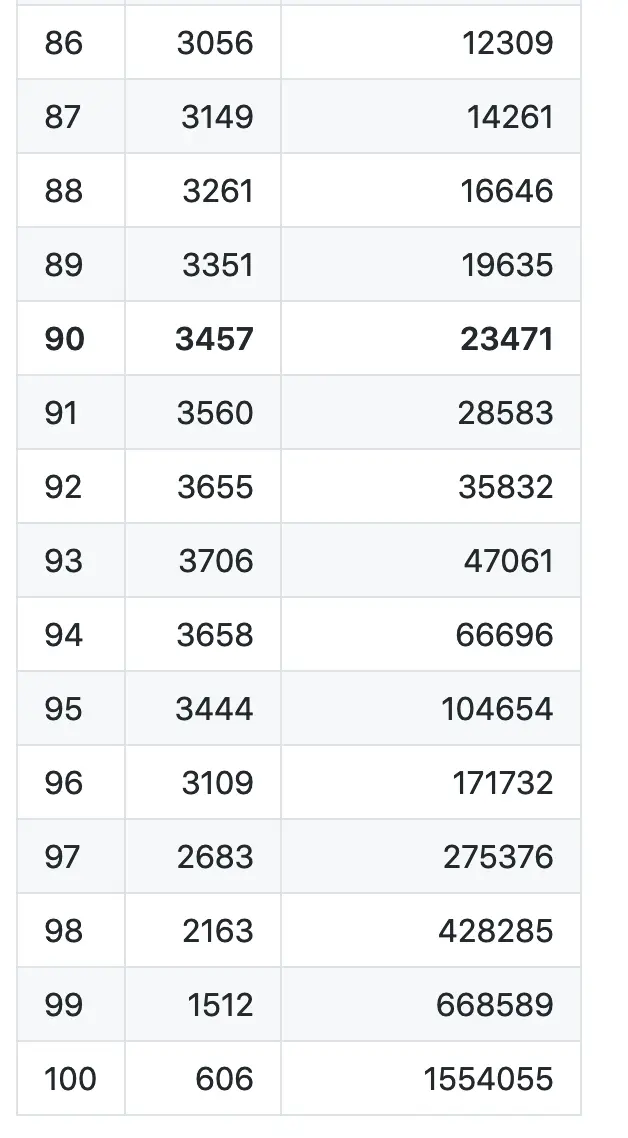
# 可以对统计结果可视化
$ TRINITY_HOME/util/misc/plot_ExN50_statistic.Rscript ExN50.stats
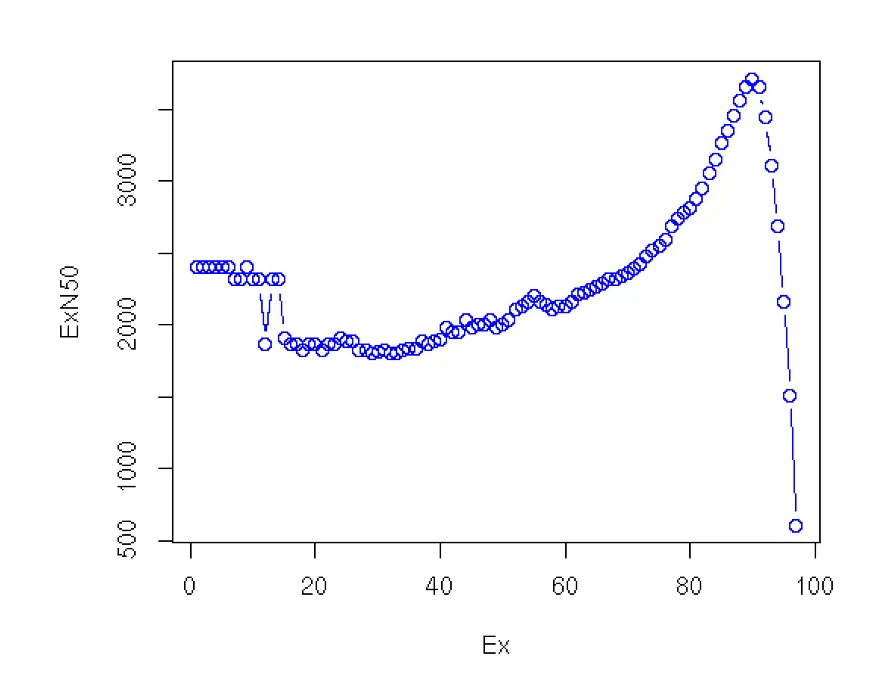
之前的统计结果N50是606 bases,图中就是100这个位置(就是因为N50计算考虑的是全部的contigs),这里其实可以过滤掉一些低表达量的contigs
可以看到总体上N50统计结果是在2.4K 到3.5K,最高值再E90的位置,我们就可以使用E90N50这个统计值(大概是3.5K)作为转录本拼接质量的评价(因为拼接约完整越好,相比于之前的606 base,是不是结果好了许多?)
参考:https://github.com/trinityrnaseq/trinityrnaseq/wiki/Transcriptome-Contig-Nx-and-ExN50-stats