097-一起来了解一下Range Data
刘小泽写于19.3.30
我们肯定都遇到许多利用坐标去处理范围信息的需求,比如要定位基因组的某个位置,这个位置可能代表了 gene model 、genetic variants(包括了SNPs、inser‐tions/deletions)、 transposable elements 、binding sites;又或者想看看染色体某个区域的GC含量、统计overlap、计算coverage、提取序列等。这些都属于Range Data的处理范围
推荐一本英文版的书 Bioinformatics Data Skills
处理Range Data一般有两种途径:R中的GenomicRanges和Linux中的bedtools
什么是RangesData?
Ranges are integer intervals that represent a subsequence of consecutive positions on a sequence like a chromosome.
它是一种坐标,记录序列信息。基因组的坐标规定都是整数,所以不会出现类似这样的坐标(50,403,503.53)
坐标有三大要素:
染色体/序列名称(因为有的基因组并没拼接完,还在scaffold或contig阶段):每个基因组都由一组染色体序列组成,我们需要指定在哪个大范围中。但是目前染色体命名没有一个标准,比如UCSC和NCBI的染色体是chr开头
chr1,而emsembl直接数字1区间:比如114,414,997 to 114,693,772,由起始位点和终止位点组成
链:因为DNA是双链,所以基因的特征信息(gene feature)可以存储在正链(positive/forward)或者负链(negative/reverse)
需要注意的是,坐标信息与参考基因组相关,因此在讨论ranges的时候要注意基因组的版本号,比如
chr15:27,754,876-27,755,076这个区间在不同版本的基因组中就表示不同信息,尤其是在和别人共享信息时可以说我得到的这个范围是基于GRCh38的,或者基于GRCh37/hg19的如果之前根据旧版本的基因组得到的坐标要迁移到新版,可以使用一些工具,比如 CrossMap 【支持BED、GFF/GTF、SAM/BAM、Wiggle、VCF文件格式在不同基因组版本之间的切换】、 NCBI Genome Remapping Service 【是一个网页工具】、 LiftOver 【基于UCSC基因组浏览器】
不同的range类型
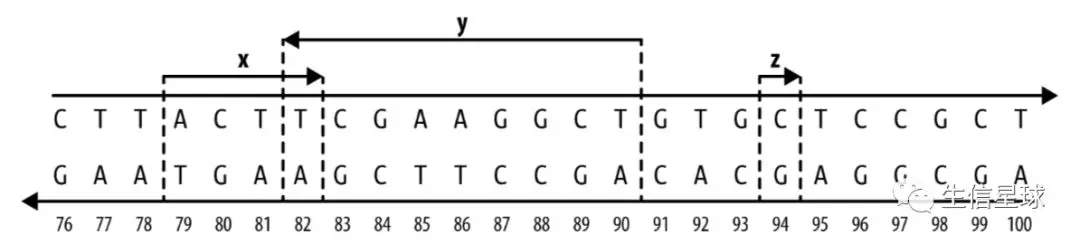
下图中x和y存在1个bp的overlap ;z没有任何overlap,只是自己跨越了1个bp;
通过箭头可以知道,x和z都是正链,分别表示ACTT和C ,y是在负链,其碱基信息就要从3’向5’看,AGCCTTCG
两套坐标基准
0-based:就是说序列的第一位坐标记为
0,最后一位坐标是序列长度-1,遵循半开半闭区间[start,end),包含左边不包含右边。例如:[1,5)表示坐标为1,2,3,4,这个模式就和Python一样>>>"CTTACTTCGAAGGCTG"[1:5] 'TTAC'1-based:这个好像比较符合我们的习惯,从1开始,遵循双闭区间
[start,end],两边都包括。例如:[2,5]就是2,3,4,5,这个模式就和R一样> substr("CTTACTTCGAAGGCTG", 2, 5) [1] "TTAC"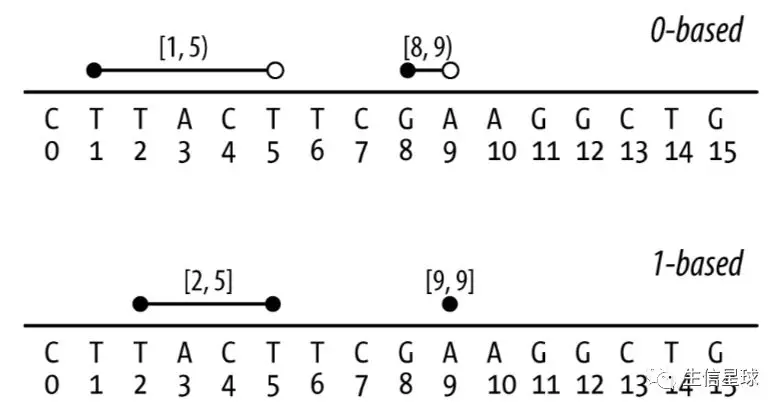
这两种系统各有优劣:
尽管我们认为1-based更符合自然计数法则,但是有些情况下并不好用,比如: 计算区间长度(range width/span)时,使用0-based系统,直接用
end-start就好,这也比较好理解;但是1-based系统需要end-start+1另外,0-based系统支持
zero-width feature,常用来描述两个碱基之间的位置,比如在上图中我们现在找一个酶切位点[12,12),然后序列就被分成了CTTACTTCGAAGG和CTG;这一点1-based系统也不能实现,它最小就是1个碱基
不同文件支持的坐标系统不同:

比较麻烦的链信息
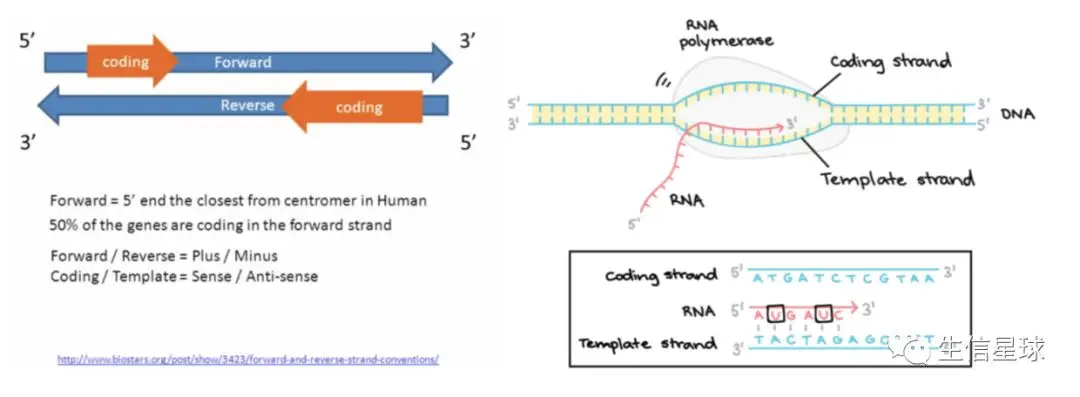
从https://www.biostars.org/p/3423/看了几个重要的概念:
- forward strand, this means reading left-to-right, and for the reverse strand it means right-to-left
- A gene can live on a DNA strand in one of two orientations. The gene is said to have a coding strand (also known as its sense strand), and a template strand (also known as its antisense strand).
- mRNA sequence always corresponds to the 5-3 coding sequence of a gene.
- mRNA matches the coding sequence of the gene, not the template sequence( 看图) 转录时基因以负链为模板链,从负链的3‘向5’转录(合成的转录本是5‘=》3’,同时与正链/编码链上对应位置的序列一致)
注意:上图中的文件(除了Blast)都是基于参考基因组生成,而参考基因组序列是以正链为基准
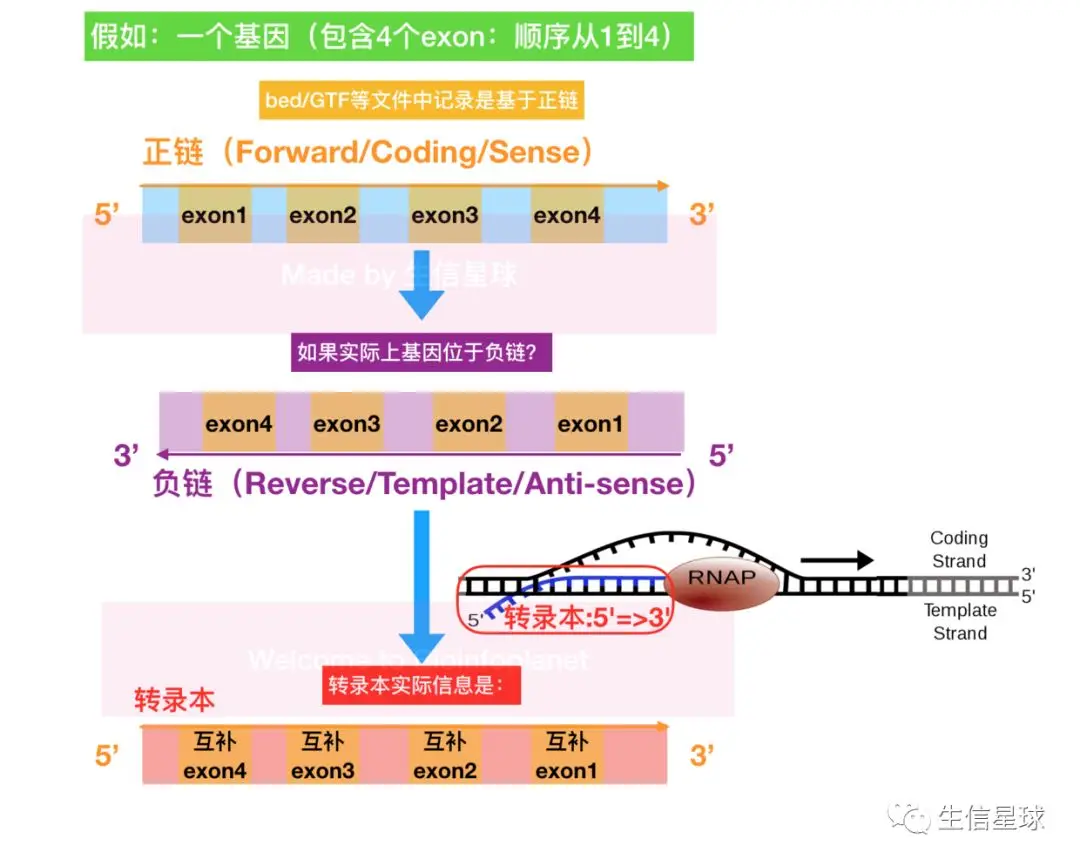
这也就是说:我们从UCSC、NCBI或Ensembl下载的参考基因组都是正链碱基序列。 但是基因分布是多样的,有的本身就在正链,即:基因对应的转录本序列恰好和正链上5‘到3’的碱基序列一致;又有的基因存在于负链,基因对应的转录本序列(以及它对应的氨基酸序列)则是和负链的5‘到3’方向的序列一致
因此如果某个基因存在于负链,从bed、GTF等文件中看到的坐标比如是chr1:2,473,087-2,492,258,抽取的这一区间序列比如是
TCTTTAC...CCGAA,其实真正NCBI记录的基因序列是TTCGG...GTAAAGA,与bed或GTF中记录的序列正好是反向互补
因此,如果想从参考基因组中抽出位于负链的基因序列,需要:1.先抽出参考基因组给的序列;2.将序列反向互补。
对于转录本在负链的情况,exon的实际位置也变了:原来在bed或GTF中forward 5'=>3’记录的第一个位置,实际上是在转录本的末尾;记录的最后一个位置
所以,我们在GTF文件中看到的正负链信息就十分有用了:
- 如果记录
+,表示在正链,那么没有问题,和文件中记录的位置和序列都一样; - 如果记录
-,那么真实信息一定是和文件记录的信息反向互补的,实际的位置也会改变
花了15分钟思考做出来这个图
最后的补充:
链的信息的确很重要,但是没有做链特异性建库时(表示我们是不知道链的方向信息的),如果要统计有多少reads比对到了某个特定的基因,一般reads会在两条链都有比对,那么这时一般会将两条链的比对结果都统计上,以免漏掉真正基因的区域