scRNA-系统学习单细胞转录组测序scRNA-Seq(六)
刘小泽写于19.4.4+4.7 结合一篇文章+scater说明书,并做了一些个人修改
首先上文章和scater包说明书
来自https://f1000research.com/articles/5-2122/v2,题目是:A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor 作者来自剑桥癌症研究所、EMBL中心、Sanger研究所,发表于2016年
文章主要借助Bioconductor包进行质量控制、数据挖掘和标准化等基本步骤,还有细胞周期判断、差异基因鉴定、降维聚类识别亚群、marker基因检测等进阶步骤。分析了几个公开可用的数据集,包括造血干细胞、脑源性细胞、辅助性T细胞和小鼠胚胎干细胞
注意:这篇文章用到的包版本比较旧,因此我们需要更新它的函数
文章中用到的主要是scater包,那么同时打开scater说明书对照看:https://bioconductor.org/packages/release/bioc/vignettes/scater/inst/doc/vignette-qc.html#cell-level-qc-metrics
分析前须知
- scRNA-seq数据比bulk RNA数据噪音更大。单个细胞中RNA含量非常低,更别说还要反转录建库测序,因此增加了Dropout(细胞中有基因却检测不到表达量)的情况
- scRNA的优势就是研究细胞的异质性。例如,识别新的细胞亚型,表征分化过程,将细胞与细胞周期阶段对应,或检测跨人群驱动差异的HVGs(highly variable genes)
- 流程从计数矩阵开始(其实上游分析也很重要,目前基本公司直接会把表达矩阵给用户,但是当结果不满意时,可以考虑上游因素)。此工作流包含了:质控(去掉有问题的细胞);标准化(细胞特异性偏差,有没有spikein等);表达矩阵探索(根据基因表达数据判断细胞周期、推断亚群)。最后,利用HVG和Marker基因对感兴趣的基因进行排序。
- 基本流程可能相同,但是不同项目中需要不同的搭配。文章使用了多个公共scRNA-seq数据集,看看如何搭配这些流程
造血干细胞分析
数据来自文章:https://f1000research.com/articles/5-2122/v2#ref-48
关键词:小鼠造血干细胞(haematopoietic stem cells ,HSCs ) 、Smart-seq2 、96孔板、ERCC、每个基因表达量由比对到外显子区域的reads数决定、 GSE61533
单细胞转录组和普通bulk转录组比对、定量流程相似,只是需要注意:
The only additional consideration is that the spike-in information must be included in the pipeline
一般来说,spike-in序列在比对之前,在基因组构建索引时是作为附加的FASTA文件;定量之前,GTF文件中也是要加上spike-in转录本和内源基因信息的
加载数据
为了更具有代表性,文章给出了读取gzip的excel文件方法。读取的表达矩阵中每一行代表一个内源基因或spike-in转录本,每一列表示一个单独的HSC细胞,然后将表达矩阵保存在 scater 的SingleCellExperiment对象中
注意:这篇文章中用到的还是scater1.0版本的
newSCESet函数,目前scater版本已经到了1.10.1,函数也升级为了SingleCellExperiment
library(R.utils)
gunzip("GSE61533_HTSEQ_count_results.xls.gz", remove=FALSE, overwrite=TRUE)
library(gdata)
# 文件24.5M,读取大概用了三分钟
counts <- read.xls('GSE61533_HTSEQ_count_results.xls', sheet=1, header=TRUE, row.names=1)
# 因为大文件读进来不容易,所以做个备份还是有帮助的
counts_bk <- counts
dim(counts)
counts[1:3,1:3]
library(scater)
sce <- SingleCellExperiment(assays = list(counts = counts),
rowData = data.frame(gene = rownames(counts)),
colData = data.frame(cell = colnames(counts)))
**看看有没有报错?**是不是提示:
Error in all_dims[, 1L] : incorrect number of dimensions想了一会,突然想起来是不是数据格式的问题,因为提示dimensions有错误,一般数据框和矩阵之间转换会出现这个问题。另外SingleCellExperiment是需要使用矩阵的(这个从它的示例数据中就可以看到)
那好,试试吧
counts <- as.matrix(counts)
sce <- SingleCellExperiment(assays = list(counts = counts),
rowData = data.frame(gene = rownames(counts)),
colData = data.frame(cell = colnames(counts)))
sce
# 这次成功了
看看行名中有没有ERCC spike-in或者MT(线粒体)基因(注意:每个数据中的线粒体缩写不一样,有的全部大写MT,有的是小写mt)
# 检测ERCC和MT,并直接统计数量
is.spike <- grepl("^ERCC", rownames(sce));sum(is.spike) # 有92个
is.mito <- grepl("^mt-", rownames(sce));sum(is.mito) # 有37个
质控
质控可以对colData中的cells和rawData中的features 分别进行统计,默认根据sce对象assays中的counts值进行质控,当存在其它表达量值比如logCounts等,可以用calculateQCMetrics的exprs_values参数进行转换
结果得到一个包含一系列统计值的,比如: total number of counts 、the proportion of counts in mitochondrial genes、spike-in transcripts等,所有统计值都储存在colData中
sce <- calculateQCMetrics(sce,
feature_controls=list(ERCC=is.spike, Mt=is.mito))
# 除了指定feature_controls,还可以指定cell_controls(比如blank wells or bulk controls)
colnames(rowData(sce))
head(colnames(colData(sce)))
结果主要有两方面:
细胞层次QC metrics,其中比较常用的统计值有:
total_counts: total number of counts for the cell (i.e., the library size)total_features_by_counts: number of features for the cell that have counts above the detection limit (default of zero)pct_counts_X:percentage of all counts that come from the feature control set named X如果我们不用counts矩阵进行计算,那么结果中所有带counts的字段都会被替换成为我们使用的矩阵的名称
基因层次QC metrics,主要包括:
mean_counts: the mean count of the gene/featurepct_dropout_by_counts: the percentage of cells with counts of zero for each genepct_counts_Y: percentage of all counts that come from the cell control set named Y(这里如果不加cell_control就没有这个结果)
输入sce可以看到,spikeNames(0)还是空值,但上面我们已经检测到了ERCC存在,因此这里我们要明确指出ERCC代表的是一个spike-in,这很重要 ,因为下游分析时需要对spike-in进行一些处理,比如方差估计和标准化。可以利用isSpike指定
# 对照组/控制条件可以根据基因(features)定,比如spike-in转录本, 线粒体基因;也可以根据细胞定,如空的测序孔、损伤的细胞等
library(SingleCellExperiment)
isSpike(sce,"ERCC") <- is.spike
# 如果要加其他的spike-in,比如adam
# isSpike(sce, "Adam") <- grepl("^Adam[0-9]", rownames(sce))
# 如果要取消spike-in
# isSpike(temp,"ERCC") <- NULL
QC图
目的都是为了帮助判断数据质量如何
检查表达量最高的一些基因(默认前50)
图中每一行是一个基因,每一个bar(线条)就是这个基因的单个细胞中的表达量,圆点是每个基因的平均表达量
plotHighestExprs(sce, exprs_values = "counts")

这个结果一般是会包含mitochondrial genes, actin, ribosomal protein, MALAT1 ,spike-in ,但是表达量前50基因中spike-in如果太多,就说明一开始加的spike-in太多;另外如果出现太多的"假基因"或者预测的基因,就说明可能比对过程出了问题。
Frequency of expression against mean
有表达基因的细胞所占的比例(纵坐标)与基因表达量平均值(横坐标)构成,对于大多数基因,X和Y坐标应该是正相关(也就是说,随着细胞数量的增加,基因表达量也是不断增加的)。如果出现负相关的离群点(outlier)【少量可以接受,但总趋势还是要正相关】,具体原因需要深入研究, 比如:**横坐标低纵坐标高的情况:**如果比对过程产生了许多"表达"的假基因,但实际上平均到所有细胞这部分就比较少(因为并不是所有细胞都会有假基因);(基因表达量的增长跟不上细胞数量的增长) 横坐标高纵坐标低的情况: PCR扩增偏差或罕见细胞群的存在,导致少数细胞中平均表达量很高(细胞数量的增长跟不上基因表达量的增长)
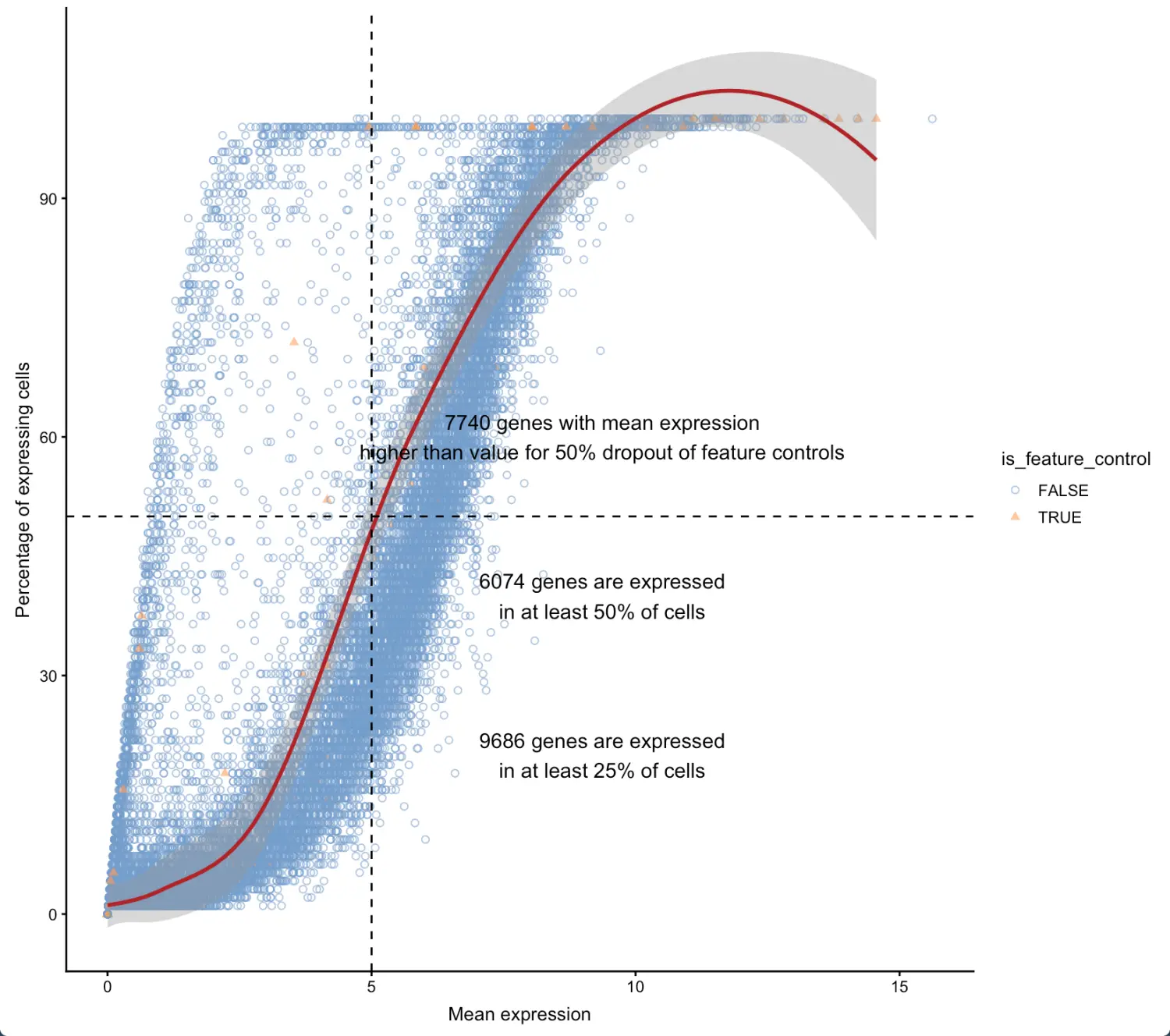
表达基因与feature controls的对比
利用colData(sce)中的纵坐标是feature control的比例,横坐标是表达的基因的数量。比较好的数据就是有大量表达的基因同时feature controls很少;如果feature controls较高而表达基因数很少,说明细胞测序失败
plotColData(sce, x = "total_features_by_counts",
y = "pct_counts_feature_control") +
theme(legend.position = "top") +
stat_smooth(method = "lm", se = FALSE, size = 2, fullrange = TRUE)
# 另外如果有分组信息的话,还可以按分组进行上色

根据基因信息(rowData)画图
> colnames(rowData(sce))
[1] "gene" "is_feature_control"
[3] "is_feature_control_ERCC" "is_feature_control_Mt"
[5] "mean_counts" "log10_mean_counts"
[7] "n_cells_by_counts" "pct_dropout_by_counts"
[9] "total_counts" "log10_total_counts"
> plotRowData(sce, x = "n_cells_by_counts", y = "mean_counts")

也可以用multiplot同时画多个图
p1 <- plotColData(sce, x = "total_counts",
y = "total_features_by_counts")
p2 <- plotColData(sce, x = "pct_counts_feature_control",
y = "total_features_by_counts")
p3 <- plotColData(sce, x = "pct_counts_feature_control",
y = "pct_counts_in_top_50_features")
multiplot(p1, p2, p3, cols = 3)

过滤之按细胞过滤
总目标:去掉不好的细胞(文库太小的)、去掉不好的基因(低表达的)、去掉线粒体占比太高的、去掉spike-in占比太高的
解决前两项问题:所有细胞中文库大小和有表达基因的数目
标准就是:总表达量(文库)小的不行,表达基因太少的也不行
先看看过滤前的原始数据
> par(mfrow=c(1,2))
> hist(sce$total_counts/1e6, xlab="Library sizes (millions)", main="",
+ breaks=20, col="grey80", ylab="Number of cells")
> hist(sce$total_features_by_counts, xlab="Number of expressed genes", main="",
+ breaks=20, col="grey80", ylab="Number of cells")

过滤的时候不能直接使用绝对值,因为可能出现这种情况:不管细胞质量如何,只要测序深度高,就能得到更多的reads。手动设置阈值往往存在把控不到位的情况,对于没有分析经验的我们来讲,使用isOutlier进行阈值设定是更好的选择,也就是先将文库的中位数进行log转换(排除太大或太小值的影响),然后减去3倍的 MAD值(median absolute deviations)。
isOutlierdefines the threshold at a certain number of median absolute deviations (MADs) away from the median
为何设定3倍的MAD? :通常把偏离中位数三倍以上的数据作为异常值,和均值标准差方法比,中位数和MAD的计算不受极端异常值的影响,结果更加稳健。
libsize.drop <- isOutlier(sce$total_counts, nmads=3, type="lower", log=TRUE)
feature.drop <- isOutlier(sce$total_features, nmads=3, type="lower", log=TRUE)
解决后两项问题:线粒体占比和spike-in占比
par(mfrow=c(1,2))
hist(sce$pct_counts_Mt, xlab="Mitochondrial proportion (%)",
ylab="Number of cells", breaks=20, main="", col="grey80")
hist(sce$pct_counts_ERCC, xlab="ERCC proportion (%)",
ylab="Number of cells", breaks=20, main="", col="grey80")

同样设置偏离中位数三倍以上的数据为离群值
mito.drop <- isOutlier(sce$pct_counts_Mt, nmads=3, type="higher")
spike.drop <- isOutlier(sce$pct_counts_ERCC, nmads=3, type="higher")
最后只筛选去掉以上四种离群值的数据:
sce <- sce[,!(libsize.drop | feature.drop | mito.drop | spike.drop)]
data.frame(ByLibSize=sum(libsize.drop), ByFeature=sum(feature.drop),
ByMito=sum(mito.drop), BySpike=sum(spike.drop), Remaining=ncol(sce))
# ByLibSize ByFeature ByMito BySpike Remaining
1 2 2 6 3 86
## 可以看到去掉上面四项内容后还有86个细胞文库
过滤后再次检测
因为我们之前做的过滤是假设所有细胞都是高质量的,但是事实上可能会有技术原因导致的低质量细胞,最好的检测办法是基于QC metrics进行PCA,检测离群点
> sce <- runPCA(sce, use_coldata = TRUE,
+ detect_outliers = TRUE)
> plotReducedDim(sce, use_dimred="PCA_coldata")
> summary(sce$outlier) # 发现还有离群点,需要去掉
Mode FALSE TRUE
logical 85 1
> sce <- sce[,-sce$outlier]
> sce <- runPCA(sce, use_coldata = TRUE,
+ detect_outliers = TRUE)
> summary(sce$outlier)
Mode FALSE
logical 85
细胞周期预测
预测的方法参考:
Scialdone et al. (2015) 利用小鼠胚胎干细胞每两个基因表达量之间进行比较,结果作为一组,最后得到
scran包中的预先训练数据集。然后基于与细胞周期相关的转录过程的保守性,利用cyclone函数对其他类型的细胞进行细胞周期预测
mm.pairs <- readRDS(system.file("exdata", "mouse_cycle_markers.rds", package="scran"))
# 除了mouse还有human的数据
library(org.Mm.eg.db)
anno <- select(org.Mm.eg.db, keys=rownames(sce), keytype="SYMBOL", column="ENSEMBL")
ensembl <- anno$ENSEMBL[match(rownames(sce), anno$SYMBOL)]
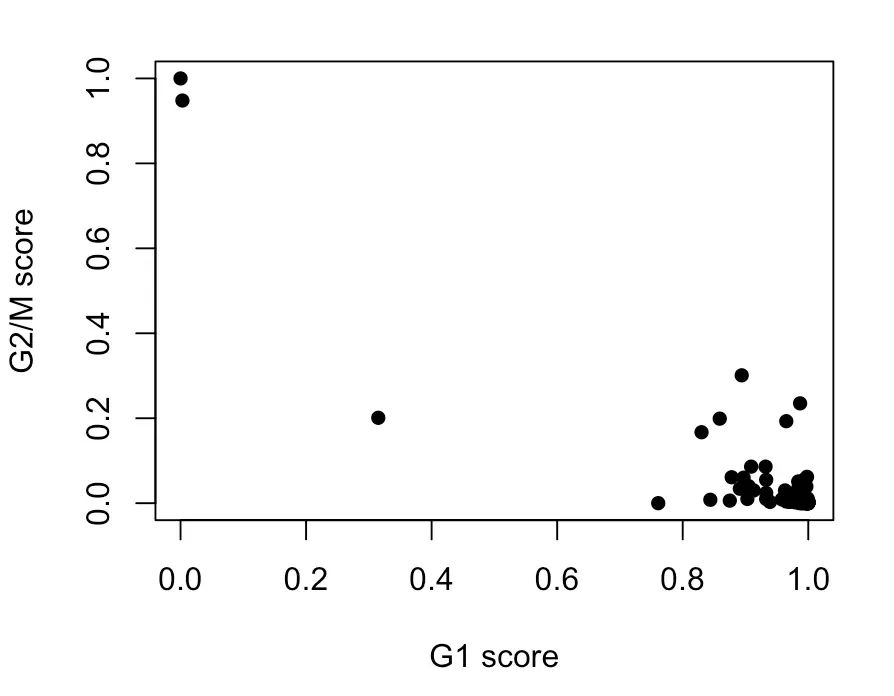
assignments <- cyclone(sce, mm.pairs, gene.names=ensembl)
plot(assignments$score$G1, assignments$score$G2M, xlab="G1 score", ylab="G2/M score", pch=16)
# 于是结果又过滤掉3个细胞
sce <- sce[,assignments$phases=="G1"]
得到的结果中,如果G1 score大于G2/M且大于0.5,就是G1期;如果G2/M大于G1且大于0.5,就是G2/M期;如果结果都不大于0.5,就是S期。[ 补充:关于细胞分期]
过滤之按基因过滤
Bourgon et al., 2010 研究表明,基因表达量为0或者接近0不能进行有效地统计分析。这里低丰度基因被定义为平均表达量低于过滤阈值1的基因,去除这些基因可以降低离散性,减少计算量,而不会造成大量信息丢失。
ave.counts <- rowMeans(counts(sce))
keep <- ave.counts >= 1
sum(keep)
单细胞特有的标准化
deconvolution方法计算标准化因子
Stegle et al., 2015 研究表明,read counts 在细胞间因捕获效率和测序深度而产生不同。在下游分析细胞间的偏差时,需要消除细胞间的这种无关生物因素的偏差(bias)。一般是先假设大部分基因在细胞间不是差异表达的,那么两个细胞间的多数非表达基因产生的count size差异就主要是由于bias产生,这部分就是需要标准化的。
每个文库中需要被标准化的counts值就是**“size factors”** 。计算size factors有多种方法,比如在bulk转录组中:利用DESeq2的 estimateSizeFactorsFromMatrix,或者edgeR的calcNormFactors。但是单细胞数据有个特点,它包含了许多的低表达量或0值,因此可以先将多个细胞的count值加起来,提高count size然后再计算总size factor,再利用deconvolved的算法得到细胞水平的factor
sce <- computeSumFactors(sce, sizes=c(20, 40, 60, 80))
summary(sizeFactors(sce))
plot(sizeFactors(sce), sce$total_counts/1e6, log="xy",
ylab="Library size (millions)", xlab="Size factor")
图中可以看到,size factors和 library sizes for all cells强相关。也就说明了细胞间的不同主要是由于捕获效率或测序深度的影响。如果是真正的细胞间差异基因,它们的total count 和size factor之间应该是一条非线性相关的趋势线,线周围是升高的散点。

对spike-in transcripts计算size factors
计算内源基因size factor的方法不适用于spike-in transcripts,试想一下没有文库定量的实验(每个文库在混合和混养测序之前的cDNA含量都不等),细胞中内源基因的RNA越多,就越需要更大的size factor去进行标准化。
但是在文库制备时,每个细胞中加入的是等量的spike-in,因此它的含量和不同文库中RNA含量无关。如果也要按照基因层面计算的size factor进行标准化,一般会导致标准化过度,表达定量不准确。
同样的思路也适用于有文库定量的情况。在cDNA总量不变的情况下,任何内源性RNA含量的增加都会抑制spike -in转录本的数目。因此, spike-in计数的bias将与基于基因的size factor得到的bias相反。
sce <- computeSpikeFactors(sce, type="ERCC", general.use=FALSE)
# general.use=FALSE保证了得到的size factor只适用于spike-in transcripts 而非内源基因
将计算的size factor用于基因表达标准化
原来的count值被用来计算标准化的log count值,下游分析基于这个。计算过程是这样的:每个细胞中的每个count值先进行log(count+1),然后除以特定细胞的size factor
sce <- normalize(sce)
标准化后,就可以探索实验因子与表达量的关系
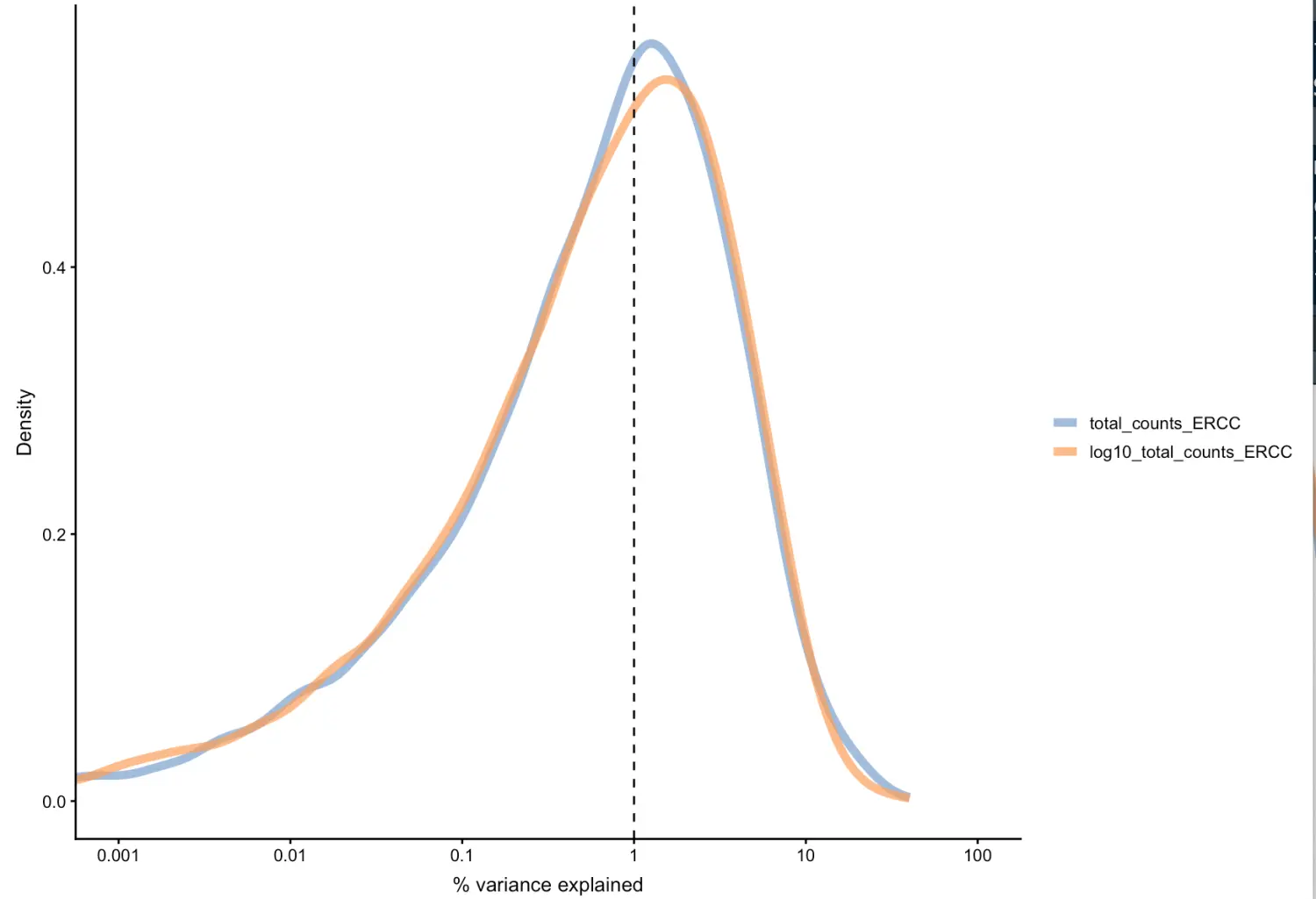
看看是否存在技术性因素对基因表达的异质性有实质性贡献。这里看下spike-in的影响
plotExplanatoryVariables(sce,
variables = c("total_counts_ERCC",
"log10_total_counts_ERCC"))