scRNA-单细胞转录组学习笔记-1
刘小泽写于19.6.10 笔记目的:根据生信技能树的单细胞转录组课程探索smart-seq2技术相关的分析技术 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=53
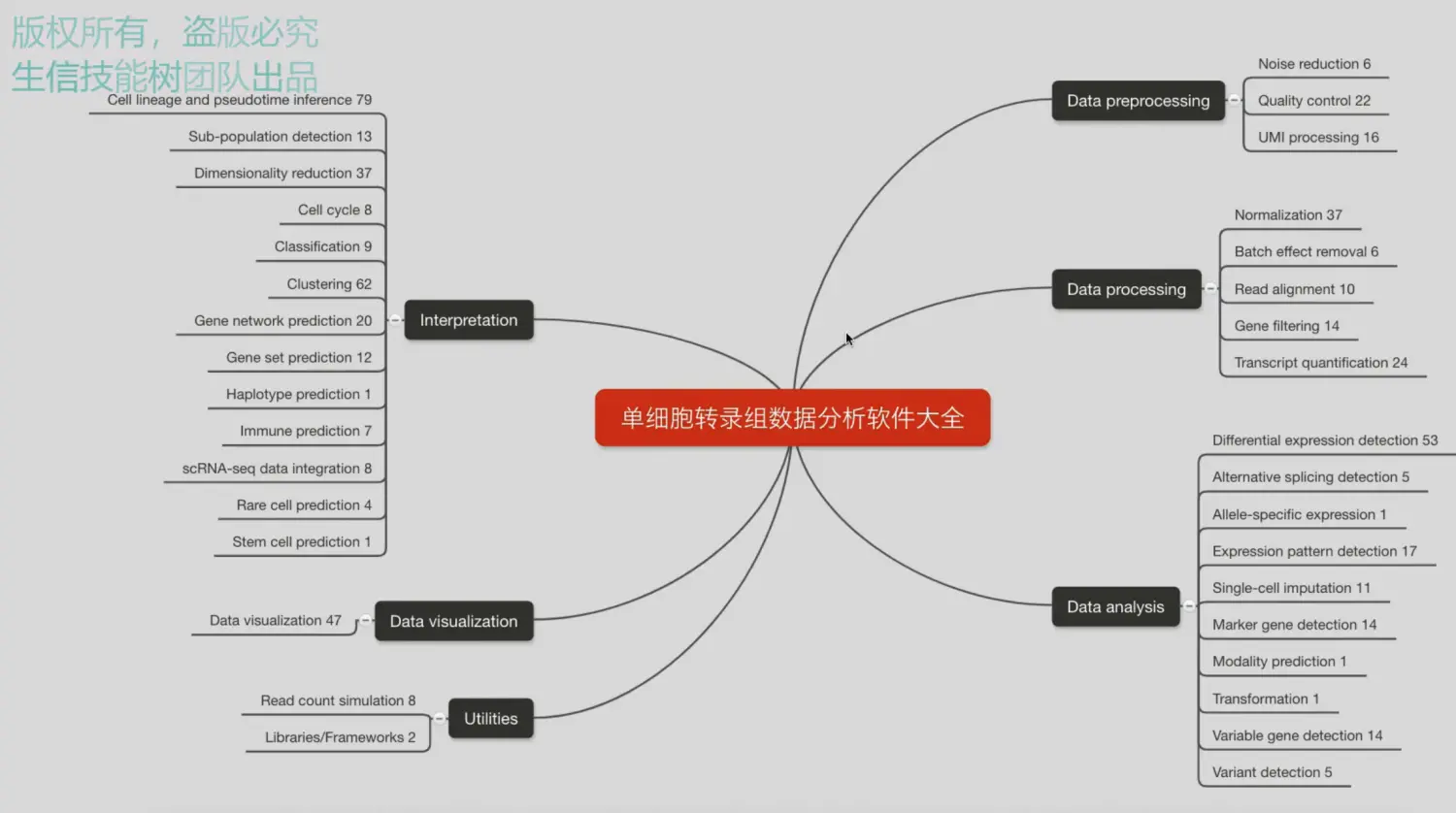
整个课程的框架
分为了5个单元,目前更新了3个单元,其中第一单元为背景介绍,包括单细胞转录组近10年的发展历程,以及两大主流技术smart-seq2(力求检测到单个细胞的基因数量)和10x(追求检测到的细胞数量)的介绍;第二单元加入了常规转录组的分析流程;第三单元重点利用smart-seq2技术得到的结果,结合三个R包进行分析;未来第4个单元将会整合公共数据库(TCGA、METABRIC)以及文献中的数据集进行整合分析;最后一部分就是展望,介绍10X的数据分析
蓬勃发展的单细胞转录组
与普通bulk转录组最大的区别就是:普通的是以一群细胞为一个样本,最后得到结果是一个均值,而单细胞将精度提高,将一个细胞作为一个样本,从而可以看出细胞的异质性
目前单细胞领域文章发表量迅速升高,有的团队一年可以出8-9篇CNS,最著名就是北大的汤富酬教授研究发育生物学领域,另外肿瘤、免疫研究相关也十分火热
不管怎样,单细胞转录组数据到了生信工程师手中,即使生物背景知识可能不完全理解,但他确实可以通过数据分析去说明某些事情。
推荐一个网站:https://omictools.com/single-cell-rna-seq-category
首先原始数据产出就有两种主流途径:测更多的细胞(以10X为主打)和测更多的基因(以smart-seq2为主打);
然后数据就要经过一系列的质控才能开始上游分析,也就是预处理阶段 ,也是两种方法:UMI、ERCC。因为单细胞精度太高,每个细胞都是独特的,和普通的Bulk RNA-seq不同,材料不容易获得,不太好做重复,因此通过生物学重复来评价技术手段/数据质量的方法不靠谱。
- UMI即unique molecular identifier,是一段随机序列,每一个DNA分子都有自己的UMI序列。可以大大降低PCR误差(比如:原来两个样本中某基因表达量相同,但是由于两个样本扩增效率不同,样本1为99%,样本2只有95%,那么同时扩增40个循环,这同一个基因就有了
0.99^40 / 0.95^40 = 5.2倍差异,因此本来没有差异也会因为外界因素扩增效率的影响而产生“假阳性”);设计不同标签的数量,大大超过待扩增的转录本,产生独特标记的分子,并允许控制扩增偏差【例如10-mer的UMI,就会有 4的十次方 约等于100万种变化】 - ERCC就是外源RNA对照联盟开发的人工设计好的已知序列和数量的mRNA,高的ERCC含量与低质量数据相关,并且通常是排除的标准
关于QC的注意点: QC不是仅仅对于下机的fq数据有效,它要在每一个分析环节都有体现。 主要包括:
- 一般的diagnostic plots: GC content, adapter, kmer, duplicated, base pair quality
- mapping reads/rate
- total sequencing reads (library size)
- the number of detected genes:检测dropouts,也就是实际有表达却没被检测到的基因
- ERCC spike-ins content
- the percentage of housekeeping genes, cell cycle genes, highly expressed genes, mitochondrial genes
- overall gene expression patterns
质控的表达矩阵要进行归一化,因为不同的细胞有不同特征,另外实验条件也不同;这对于解释单细胞数据是至关重要的。单细胞的样本量相比bulk实在小太多,因此更容易引入技术噪音,这个必须去除;
加入了降维这重要的一步,因为细胞数量太多,也就是要分析的样本数量要几百甚至几千,于是会产生几百或几千的维度(可以试想一下常见的三维、四维空间);
找差异基因、找marker基因(和差异基因不同,它不需要有差异,只需要有重要的生物学意义)、根据基因重要性进行细胞分群
看文献
构建文库
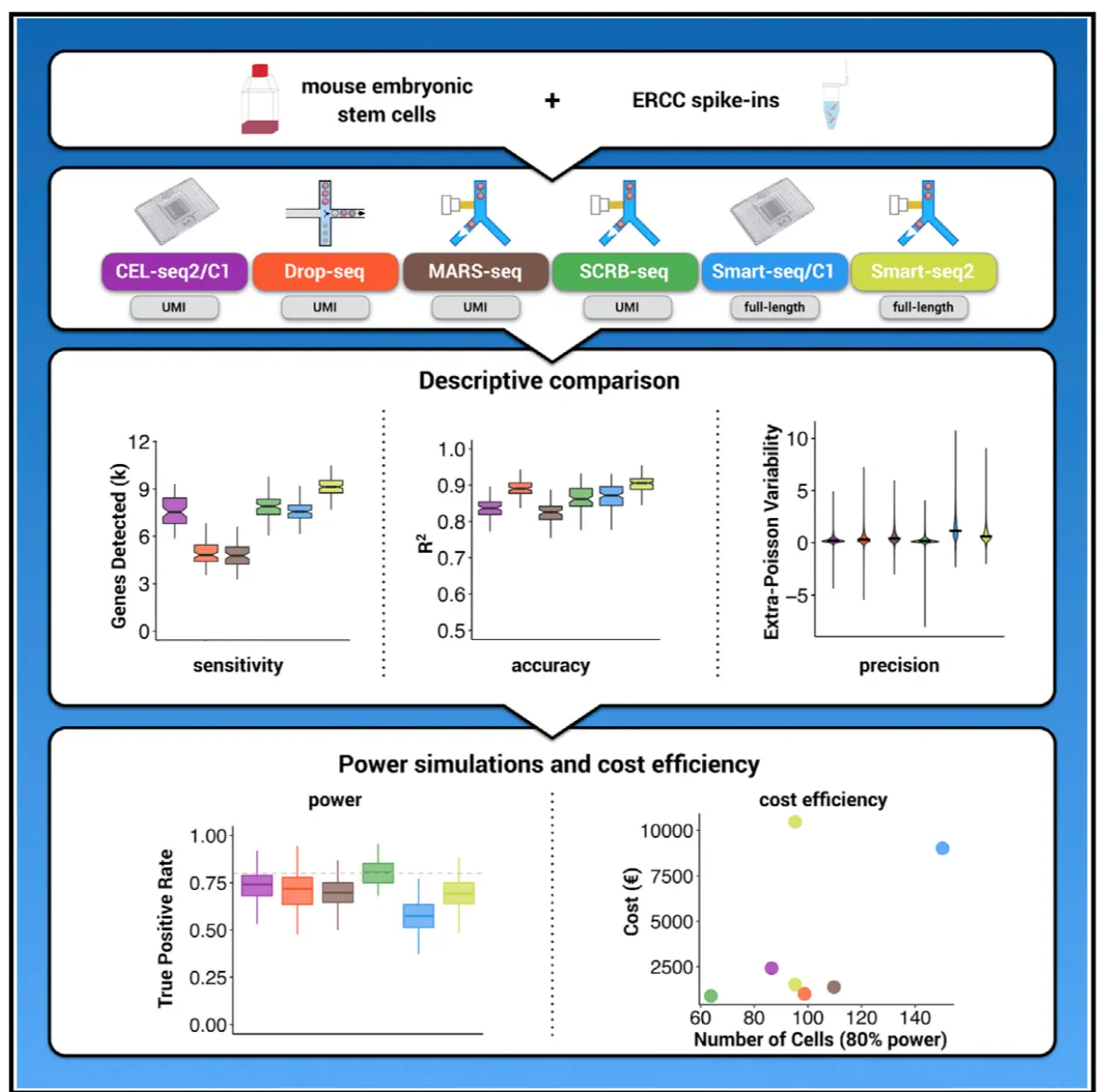
综述:Comparative Analysis of Single-Cell RNA Sequencing Methods. 2017, (doi: 10.1016/j.molcel.2017.01.023.)
涉及到了6中文库构建方法(CEL-seq2, Drop-seq, MARS-seq, SCRB- seq, Smart-seq, and Smart-seq2),可以再结合相关的每一个文库找6篇文章 文章发现:Smart-seq2可以在每个细胞中找到最多的基因,同样费用比较高;检测少量细胞时,MARS-seq、SCRB-seq、Smart-seq2更有效
归一化
文献1:Assessment of Single Cell RNA-Seq Normalization Methods,2017 (doi: 10.1534/g3.117.040683)
评价了几种归一化方法:
- fragments per kilobase of transcript per million mapped reads (FPKM)(Mortazavi et al., 2008)
- upper quartile (UQ)(Bullard et al., 2010)
- Trimmed mean of M-values (TMM)(Robinson and Oshlack, 2010)
- DESeq(Love et al.,2014)
- removed unwanted variation (RUV)(Risso et al., 2014)
- gamma regression model (GRM)(Ding et al., 2015).
文献2:Performance Assessment and Selection of Normalization Procedures for Single-Cell RNA-Seq, 2019 (DOI:https://doi.org/10.1016/j.cels.2019.03.010)
主要研究了scone方法:a flexible framework for assessing performance based on a comprehensive panel of data-driven metrics
(http://bioconductor.org/packages/scone/)
另外方法还有很多,比如:LSF(Lun Sum Factors),BigNorm, Scnorm, BASiCS, RLE(size factor relative log expression)
降维
PDF: https://lib.ugent.be/fulltxt/RUG01/002/349/740/RUG01-002349740_2017_0001_AC.pdf 值得好好阅读,讲了许多关于降维原理和应用的知识
文中1.5.1部分(Clustering high-dimension to identify subtypes)写出:
Importantly, the reduced dimensionality data are less noisy than the high-dimensional data bust lose some of the biological variance.
文章1: PCA, MDS, k-means, Hierarchical clustering and heatmap.
文章2: Outlier Preservation by Dimensionality Reduction Techniques “MDS best choice for preserving outliers, PCA for variance, & T-SNE for clusters”
鉴定细胞群
每个术语都对应一篇文献
- 降维:PCA、tSNE、DM(Diffusion maps)
- feature selection:M3Drop(Michaelis-Menten Modelling of Dropouts)、HVG(Highly variable genes)、Spike-in based methods、Correalated expression
- Seurat:is an R package designed for the analysis and visualization of single cell RNA-seq data. It contains easy-to-use implementations of commonly used analytical techniques, including the identification of highly variable genes, dimensionality reduction (PCA, ICA, t-SNE), standard unsupervised clustering algorithms (density clustering, hierarchical clustering, k-means), and the discovery of differentially expressed genes and markers.
- SC3:SC3 achieves high accuracy and robustness by consistently integrating different clustering solutions through a consensus approach. Tests on twelve published datasets show that SC3 outperforms five existing methods while remaining scalable, as shown by the analysis of a large dataset containing 44,808 cells. Moreover, an interactive graphical implementation makes SC3 accessible to a wide audience of users, and SC3 aids biological interpretation by identifying marker genes, differentially expressed genes and outlier cells.
- tSNE+kmeans
- SNN-Clip: doi: 10.1093/bioinformatics/btv088
- SINCERA: SINCERA: A Pipeline for Single-Cell RNA-Seq Profiling Analysis.
综述: A systematic performance evaluation of clustering methods for single-cell RNA-seq data (SC3 and Seurat show the most favorable results)
关于各种单细胞工具:https://www.scrna-tools.org/ 文章在: Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database