scRNA-单细胞转录组学习笔记-4
刘小泽写于19.6.14-15 笔记目的:根据生信技能树的单细胞转录组课程探索smart-seq2技术相关的分析技术 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=53 第二单元第二、三讲:获取Github代码包以及准备工作
先下载代码包
github代码在:https://github.com/jmzeng1314/scRNA_smart_seq2/archive/master.zip
首先进入RNA-seq目录,从step0-step9是对常规转录组的一个回顾
准备工作之R包
从step0开始,代码注释蛮详细的,我会挑选重要的部分写到这里,其他可以自行看代码学习,下面就是主要利用Rstudio进行操作了
一个好习惯,做新项目时记得清空之前的变量,使用
rm(list = ls())有的R包比较大,经常需要加载其他的动态库dynamically loaded libraries (DLLs),例如:
> length(loadedNamespaces()) [1] 34 > library(Seurat) #加载一个seurat包会出现接近60个依赖的动态库 > length(loadedNamespaces()) [1] 96如果不设置,就会因为加载数量超限制而报错(https://developer.r-project.org/Blog/public/2018/03/23/maximum-number-of-dlls/)
在R3.3版本中,只能有100个固定的动态库限制,到了3.4版本以后,就能够使用
Sys.setenv(R_MAX_NUM_DLLS=xxx)进行设置,而这个数字根据个人情况设定在新建数据框时会自动将字符串的列当做是因子型向量,但是我们常常还需要对字符进行修改,因此需要先将这个设置取消:
options(stringsAsFactors = F)因为Bioconductor下载方法的变动,要学会使用
BiocManager::install这个命令,例如:BiocManager::install(c( 'scran'),ask = F,update = F),新加的两个选项表示:不要问我要不要下载,直接下!还有不要问我要不要更新,不更新!【除非不升级就报错】下载包存在网络的限制,毕竟R语言是国外开发,因此可以通过
options()$repos看看常规CRAN安装R包的使用镜像(一般情况下是rstudio公司的),但是这里我们可以自行设置:比如设置成清华源:options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/")),另外Bioconductor也有自己的镜像设置:修改一下options即可,options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")# 总结一下,可以先用if判断再进行设置 if(length(getOption("CRAN"))==0) options(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/") if(!require("BiocManager")) install.packages("BiocManager",update = F,ask = F) if(length(getOption("BioC_mirror"))==0) options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")如何使用if判断语句进行包的安装:
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
最后,就是安装所有必备的R包(包括CRAN和Bioconductor)
# 快速安装cran包
cran_pkgs <- c("ggfortify","FactoMineR","factoextra")
for (pkg in cran_pkgs){
if (! require(pkg,character.only=T) ) {
install.packages(pkg,ask = F,update = F)
require(pkg,character.only=T)
}
}
# Bioconductor包
library(BiocManager)
bioc_pkgs <- c("scran","TxDb.Mmusculus.UCSC.mm10.knownGene","org.Mm.eg.db","genefu","org.Hs.eg.db","TxDb.Hsapiens.UCSC.hg38.knownGene")
for (pkg in bioc_pkgs){
if (! require(pkg,character.only=T) ) {
BiocManager::install(pkg,ask = F,update = F)
require(pkg,character.only=T)
}
}
目的:利用R包重复文章的基因数量图、聚类图、基因在聚类图中的热图、每个基因表达量在不同cluster的小提琴图
准备工作之表达矩阵

看到文章中有两个表达矩阵,其中第一个是原始表达矩阵(均为整数),第二个是rpkm是表达量归一化后的值(包含了小数),因此也能说明为何第二个文件比第一个要大。
RPKM这个指标可以这样理解:R表示reads,K表示基因长度,M表示文库大小,它实际上做的事情也就是去掉基因长度和测序文库的差异对reads比对数量的影响
好,先说说为什么要去掉文库大小差异:以这篇文章中的图片为例:https://sci-hub.tw/https://doi.org/10.1186/s13059-018-1466-5
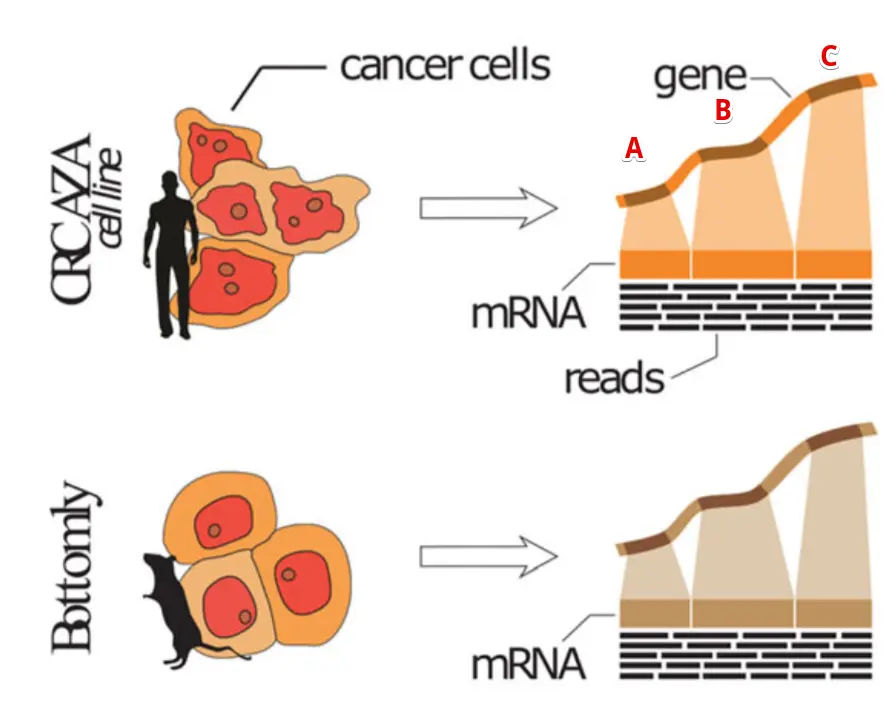
比如有两个样本,要比较三个基因ABC的表达量,图中越高表示比对到这个基因的reads数越多,因此在同一个样本中可以看到C>B>A,但是不同的两个样本呢?
测序量不同,比大小是不公平的
举个夸张的例子:上面👆的样本(简称"样本1”)中一共比对了100万条reads,其中C基因比对到了100条;下面👇的样本(简称"样本2”)中一共比对了100条reads,其中C基因比对了10条。虽然最终的数据显示:样本1中C基因比样本2的C基因比对reads数多了90条,但是考虑到实际样本情况就是,样本2中C基因可是占据了总比对量的十分之一,而样本1呢?很小很小…。因此去掉M(也就是每个样本的测序文库大小,以Million为单位)的影响,才是比较客观的。
同样的,有的基因长,有的基因短,开发RPKM的人就想:基因长的比对到的reads也会更多,因此也去掉了这个差异(除以K)
但是!这个概念目前在统计上是错误的,因此并不建议使用这个指标
操作表达矩阵
读取
# 保留头信息,并设置分隔符为制表符tab
a=read.table('../GSE111229_Mammary_Tumor_fibroblasts_768samples_rawCounts.txt.gz',header = T ,sep = '\t')
# 读进来以后,简单查看一下
a[1:6,1:4]
过滤
可以看到很多基因对应的表达量都是0

下面会用到循环,但是为了方便理解,先拿其中一行为例:
x=a[1,] #比如将第一行提取出来赋值给x
# 将x中的值与1作比较(利用了R语言的循环补齐,也就是说,它会将768个值一个一个去和1做比较,然后返回逻辑值TRUE或者FALSE)
x>1
# 然后利用table()函数检查x中有多少是TRUE,多少是FALSE
table(x>1)
# FALSE TRUE
# 766 2
# 可以看到第一行这个基因在768个细胞中只有两个细胞有表达,我们认为:这两个细胞也不好分组,cluster聚类也没有什么意义,因此可以去掉
# 但是这个细胞量设置成多少合适呢?总不能不能一股脑全设成2吧
floor(ncol(a)/50) # 用总列数除以50然后向下取整,结果就是15
# 也就是说,只要一行中至少要在15个样本中有表达量
# 上面知道了 x>1 返回逻辑值0和1,0为FALSE,1为TRUE。现在我们要找一行中总共有多少TRUE,就用sum计算一下(因为会忽略掉0的影响)
sum(x>1) > floor(ncol(a)/50)
# 当然第一行会返回FALSE,也就表明我们要去掉这一行内容
a[sum(x>1) > floor(ncol(a)/50),]# 就把不符合要求的第一行去掉了
上面,我们对一行的筛选与过滤有了认识,那么一个表达矩阵有2万多行,怎样实现循环操作呢?
# 专业的事情交给专业的工具去处理=》apply
# 要使用apply函数先要明白三个问题:对谁进行操作?对行还是列进行操作?操作什么?
apply(a, 1, function(x) {sum(x>1) > floor(ncol(a)/50)})
# 1:对a这个矩阵进行操作
# 2:对行(也就是1表示)进行操作[补充:如果对列操作,用2表示]
# 3:操作什么?复杂的操作先写上 function(x){},这是一个标准格式,然后大括号中是要进行操作的函数,于是我们就可以将我们之前写的那一行粘到这里,最后仍然是逻辑值
最后,有多少行就会返回多少个apply判断的逻辑值,显示FALSE的就是要过滤掉的,于是再用行筛选完成整个操作,并赋值给一个新变量:
dat=a[apply(a,1, function(x) sum(x>1) > floor(ncol(a)/50)),]
dim(dat)
# 12198 768 最终就保留了12198个基因
其实原文保留的更少,原文只有10835个基因