scRNA-单细胞转录组学习笔记-13-差异分析及KEGG注释简介
刘小泽写于19.8.7-第二单元最后一讲:差异分析及KEGG注释简介 笔记目的:根据生信技能树的单细胞转录组课程探索smart-seq2技术相关的分析技术 课程链接在:http://jm.grazy.cn/index/mulitcourse/detail.html?cid=53
原来的bulk-RNA差异分析一般需要比较处理组(例如有三个样本)和处理组(例如也有三个样本),这里对于单细胞来讲,每个细胞就是一个样本,于是有768个样本,但是还是不能直接进行差异分析,还是需要先分个组,看看哪些细胞离得更近,就划分为一组,最后对每个组进行比较
于是单细胞的差异分析在进行之前,就要先探索一下文章数据如何进行的分组
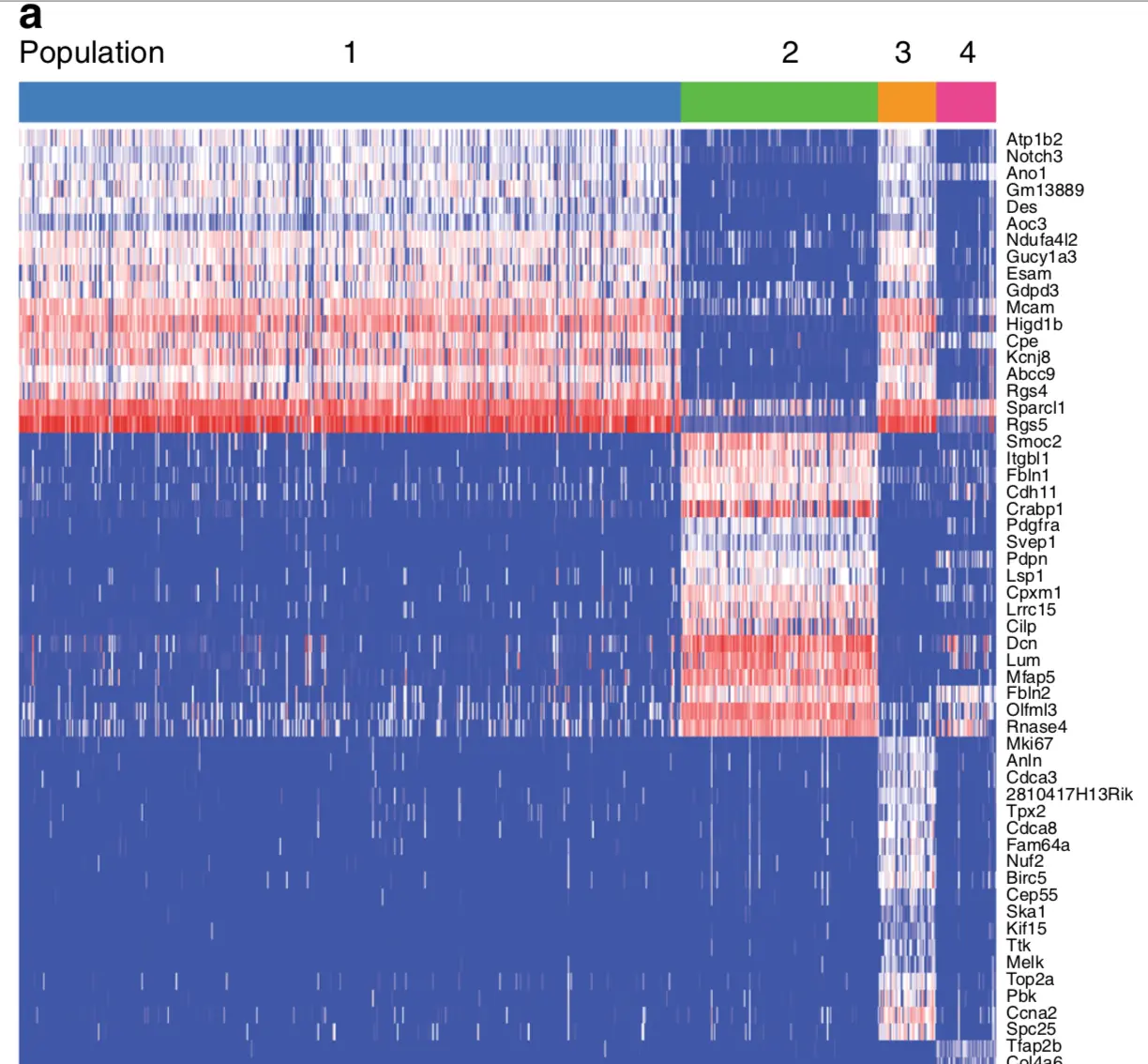
探索分组
主要利用三种方法:logCPM、RPKM、logRPKM
# 对logCPM矩阵进行分组
load(file = '../input.Rdata')
dat[1:4,1:4] ## 在step0-index.R中对原始表达矩阵进行了过滤和CPM处理:dat=log2(edgeR::cpm(dat)+1) ,将结果保存在了input.Rdata中
hc.logCPM=hclust(dist(t( dat )))
load(file = '../input_rpkm.Rdata')
# 对logRPKM矩阵进行分组
hc.logRPKM=hclust(dist(t(log(dat+0.1))))
# 对RPKM矩阵进行分组
hc.RPKM=hclust(dist(t( dat )))
# 因为三个hclust处理较慢,因此运行完可以保存为一个Rdata(另外Github下载的代码目录中是包含这个hc_3.Rdata的,如果不想自己运行,可以直接加载)
# save(hc.logCPM,hc.logRPKM,hc.RPKM,file = 'hc_3.Rdata')
load(file = 'hc_3.Rdata')
好,首先进行一下可视化
par(mfrow=c(1,3))
plot(hc.logRPKM,labels = F)
plot(hc.RPKM,labels = F)
plot(hc.logCPM,labels = F)
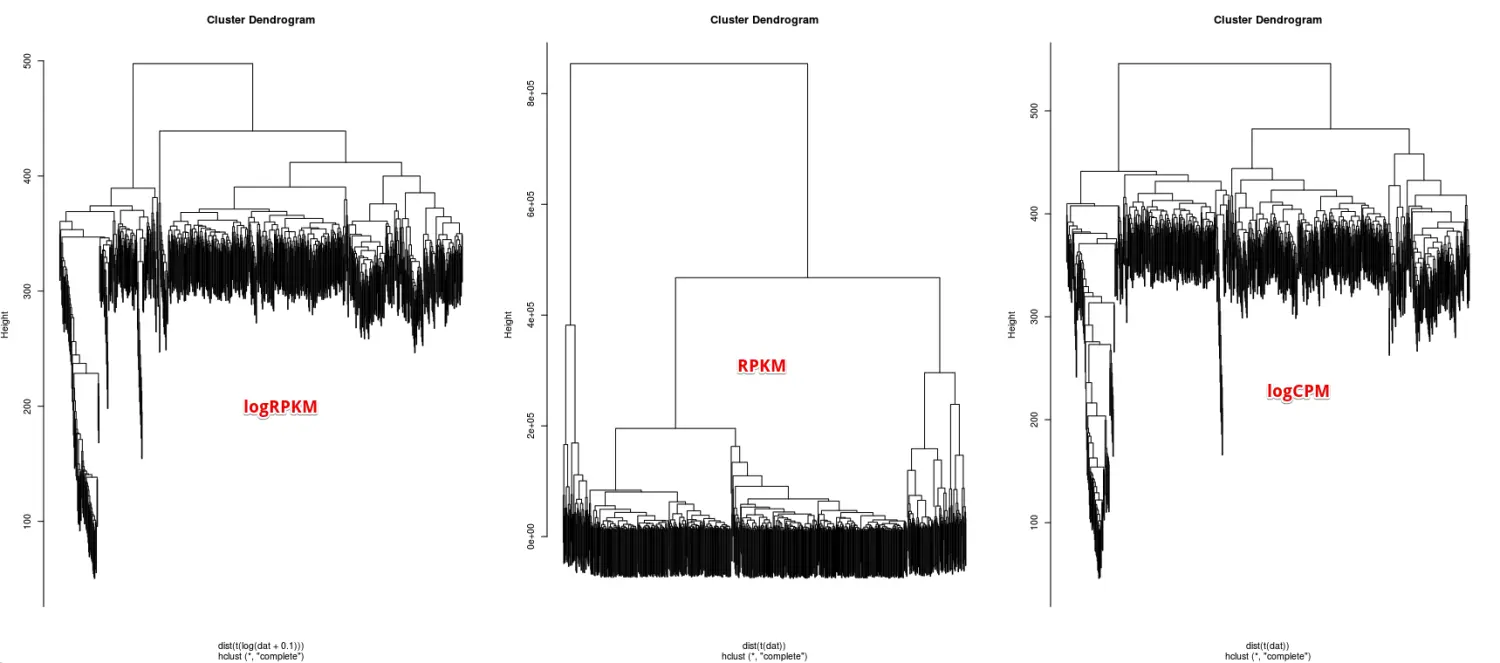
看似第一个(logRPKM)和第三个(logCPM)形状比较像,具体可以使用table()函数看一看
> g1 = cutree(hc.logRPKM, 4);table(g1)
g1
1 2 3 4
344 189 217 18
> g2 = cutree(hc.RPKM, 4) ;table(g2)
g2
1 2 3 4
112 606 15 35
> g3 = cutree(hc.logCPM, 4) ;table(g3)
g3
1 2 3 4
312 300 121 35
看顶部原文的分组图片,可以看到有一组特别多,其他三组较少,其实很像我们这里用RPKM(第二组)得到的结果,但是这个结果到底如何?我们接下来会利用tSNE方法继续判断
注意:Rtsne函数是对行进行操作,因此我们原来的表达矩阵需要转置后运行,来满足我们对样本聚类的需求。如果不确定,可以试试使用标准的表达矩阵(行为12689个基因,列为768个样本),它画出来的结果是惨不忍睹的,另外可以通过运算时间来判断,如果运行时间非常长,就要考虑:是不是矩阵没转置?
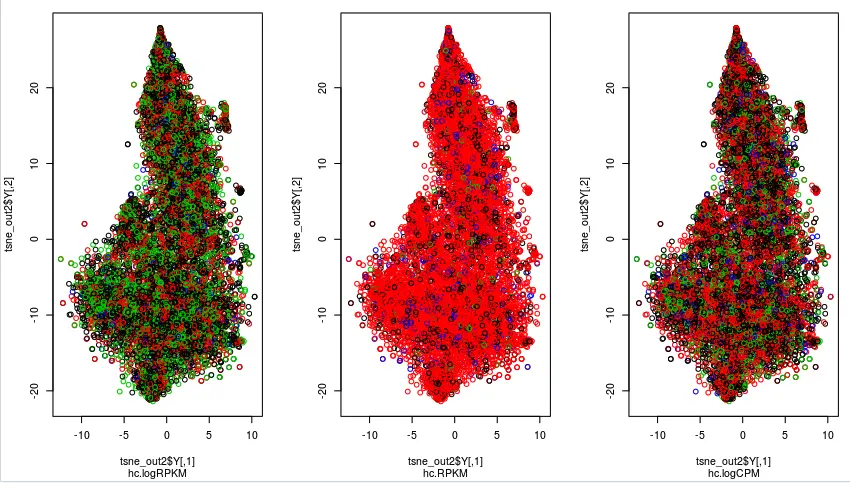
正确的做法如下:
load(file = '../input_rpkm.Rdata')
> dat[1:4,1:4] # 时刻不忘检查
SS2_15_0048_A3 SS2_15_0048_A6 SS2_15_0048_A5 SS2_15_0048_A4
0610007P14Rik 0 0 74.95064 69.326130
0610009B22Rik 0 0 0.00000 0.000000
0610009L18Rik 0 0 0.00000 0.000000
0610009O20Rik 0 0 2.19008 3.314831
dat_matrix <- t(dat) # 矩阵转置
dat_matrix =log2(dat_matrix+0.01)
set.seed(42) #因为tsne函数每次运行都会绘制新的坐标,因此需要设定随机种子,保证重复性
tsne_out <- Rtsne(dat_matrix,pca=FALSE,
perplexity=27,theta=0.5)
par(mfrow=c(1,3))
plot(tsne_out$Y,col= g1,sub = 'hc.logRPKM')
plot(tsne_out$Y,col= g2,sub = 'hc.RPKM')
plot(tsne_out$Y,col= g3,sub = 'hc.logCPM')
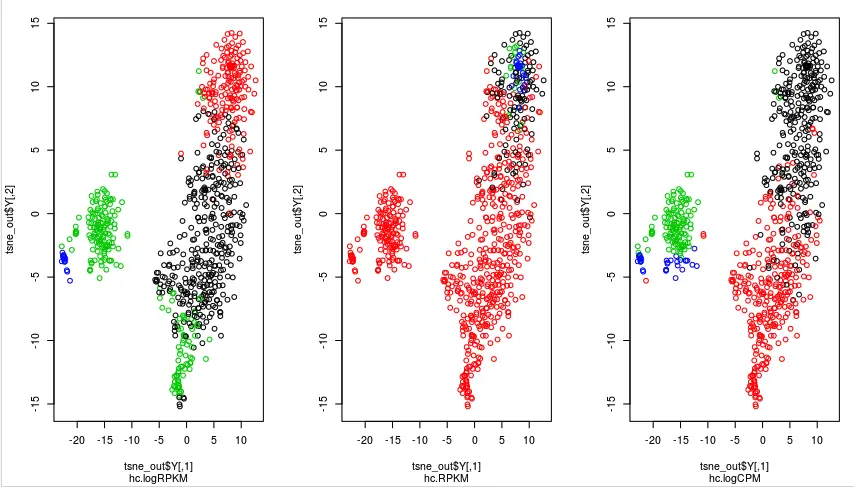
从上面👆这个图可以看到,logCPM(最右边的图)中样本分的还很开,logRPKM(最左边)次之,它把绿色的样本混在了一起,最差的是直接用RPKM得到的分组
如果使用logCPM矩阵进行作图:
load(file = '../input.Rdata')
> dat[1:4,1:4]
SS2_15_0048_A3 SS2_15_0048_A6 SS2_15_0048_A5 SS2_15_0048_A4
0610007P14Rik 0 0 6.459884 6.313884
0610009B22Rik 0 0 0.000000 0.000000
0610009L18Rik 0 0 0.000000 0.000000
0610009O20Rik 0 0 2.544699 3.025273
dat_matrix <- t(dat)
set.seed(42)
tsne_out <- Rtsne(dat_matrix,pca=FALSE,
perplexity=27,theta=0.5)
par(mfrow=c(1,3))
plot(tsne_out$Y,col= g1,sub = 'hc.logRPKM')
plot(tsne_out$Y,col= g2,sub = 'hc.RPKM')
plot(tsne_out$Y,col= g3,sub = 'hc.logCPM')
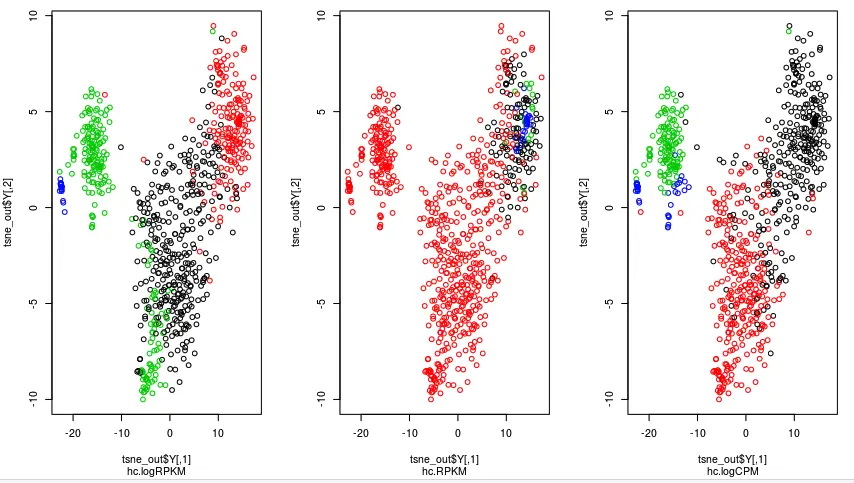
那有没有可能是因为数据计算方法不同而导致结果可视化的差异呢?也就是说,前面两个图都是使用的log矩阵,一个是logRPKM,一个是logCPM,当然它们的结果会更"偏袒"于自身,而不是原始的RPKM值。这里,可以看看直接使用RPKM矩阵进行分组作图,结果是不是RPKM分的更开(猜想应该是RPKM矩阵得到的图会更偏向于RPKM的分组)
load(file = '../input_rpkm.Rdata')
dat[1:4,1:4]
dat_matrix <- t(dat) # 这里就不再进行log处理了
set.seed(42)
tsne_out <- Rtsne(dat_matrix,pca=FALSE,
perplexity=27,theta=0.5)
par(mfrow=c(1,3))
plot(tsne_out$Y,col= g1,sub = 'hc.logRPKM')
plot(tsne_out$Y,col= g2,sub = 'hc.RPKM')
plot(tsne_out$Y,col= g3,sub = 'hc.logCPM')
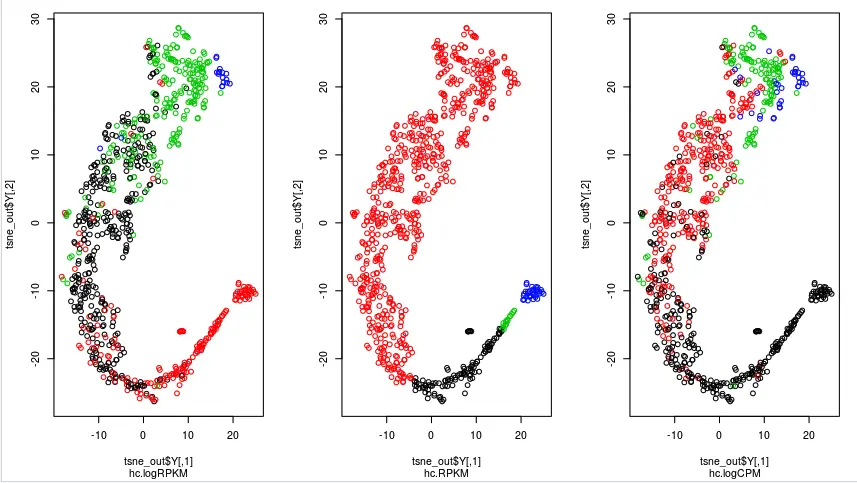
结果看到,这次RPKM分的更开了
但是,和原文不同,我们下面👇的差异分析操作中,还是更偏向于使用CPM的结果
附:运行得到的tsne_out结果是一个列表,主要包含:

差异分析
rm(list = ls())
options(stringsAsFactors = F)
load(file = '../input.Rdata')
# a[1:4,1:4]
# head(df)
> table(df$g)
1 2 3 4
312 300 121 35
接下来将使用RNA-seq常用的limma包进行处理(注意:这里只是在探索如何进行分析,单细胞数据不一定会使用到常规的差异分析包,会有更好的算法等待着我们)
如何得到差异基因?
# 对基因进行过滤(之所以赋值给exprSet,是因为下面limma运算过程中全程都在用这个名称,运行函数就是拷贝代码=》用到哪里改哪里)
exprSet=a[apply(a,1, function(x) sum(x>1) > floor(ncol(a)/50)),]
# 然后因为是smart-seq2的数据,会有ERCC spike-in,检查一下
> grep('ERCC',rownames(exprSet))
[1] 12139 12140 12141 12142 12143 12144 12145 12146 12147 12148 12149 12150 12151 12152 12153 12154
[17] 12155 12156 12157 12158 12159 12160 12161 12162 12163 12164 12165 12166 12167 12168 12169 12170
[33] 12171 12172 12173 12174 12175 12176 12177 12178 12179 12180 12181 12182 12183 12184 12185 12186
[49] 12187 12188 12189 12190 12191 12192 12193 12194 12195 12196 12197 12198
# 嗯,确实存在,然后我们要去掉,两种方法都可以(grepl返回逻辑值,grep返回坐标)
# 方法一:
exprSet=exprSet[!grepl('ERCC',rownames(exprSet)),]
# 方法二:
exprSet=exprSet[-grep('ERCC',rownames(exprSet)),]
然后limma包需要定义分组信息,这一点就要去原文寻找,他们怎么做的分组,看到:他们是将每个population和其他的混合群体进行差异分析

于是我们可以对第一组和其他组(第2、3、4组的混合)进行差异分析
# 定义分组信息
group_list=ifelse(df$g==1,'me','other');table(group_list)
# group_list
# me other
# 312 456
# 接下来直接拷贝代码,运行即可
library(edgeR)
if(T){
suppressMessages(library(limma))
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(exprSet)
design
dge <- DGEList(counts=exprSet)
dge <- calcNormFactors(dge)
logCPM <- cpm(dge, log=TRUE, prior.count=3)
v <- voom(dge,design,plot=TRUE, normalize="quantile")
fit <- lmFit(v, design)
group_list
cont.matrix=makeContrasts(contrasts=c('me-other'),levels = design)
fit2=contrasts.fit(fit,cont.matrix)
fit2=eBayes(fit2)
tempOutput = topTable(fit2, coef='me-other', n=Inf)
DEG_limma_voom = na.omit(tempOutput)
head(DEG_limma_voom)
}
# 部分结果在此
logFC AveExpr t P.Value adj.P.Val B
Prelid1 -4.099869 6.527634 -28.15400 1.120169e-120 1.359661e-116 264.7042
Sec14l4 5.251698 4.059171 26.42578 3.212438e-110 1.949628e-106 240.8450
Shank1 5.274392 4.317943 26.17762 1.015632e-108 4.109248e-105 237.3548
Jph3 4.994536 3.689781 25.66937 1.190962e-105 3.613976e-102 230.3916
Atp5f1 -4.088563 6.776594 -25.17238 1.181824e-102 2.868996e-99 223.4194
Sec13 -4.177628 6.377070 -24.09519 3.515681e-96 7.112222e-93 208.6103
看到在第一组中有个基因Sec14l4相对于其他组(第2、3、4)是高表达的 (logFC=5.251698) ,于可以可视化一下,最直观的就是利用箱线图,然后加上分组信息即可
boxplot(dat['Sec14l4',]~df$g)
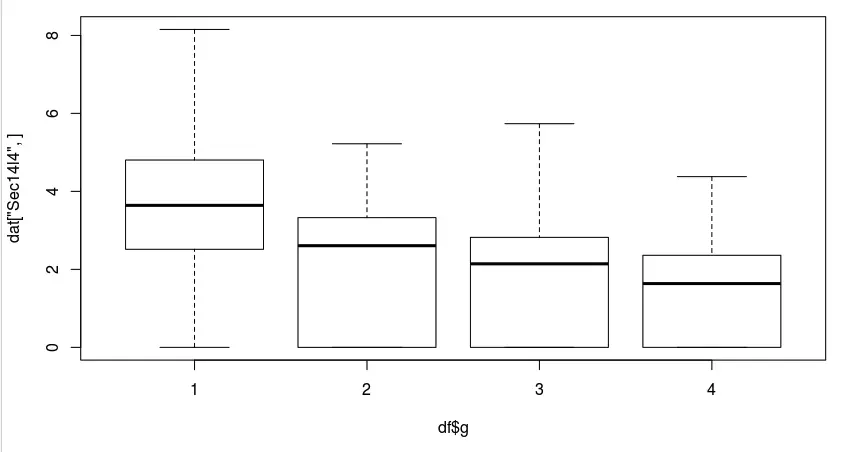
那么好,现在处理完了第一组和其他组的比较,得到的在第一组的差异基因,那么如何得到其他组中的差异基因呢?最先想到的会是将上面👆的代码
group_list=ifelse(df$g==1,'me','other')这里改一下,然后再统统运行一遍。这样是可以的,但是代码发生了大规模重复,说明有问题,不美观也不具备可重复性。解决方法就是:写函数
# 定义一个函数,输入是exprSet和group_list
do_limma_RNAseq <- function(exprSet,group_list){
suppressMessages(library(limma))
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(exprSet)
design
dge <- DGEList(counts=exprSet)
dge <- calcNormFactors(dge)
logCPM <- cpm(dge, log=TRUE, prior.count=3)
v <- voom(dge,design,plot=TRUE, normalize="quantile")
fit <- lmFit(v, design)
group_list
cont.matrix=makeContrasts(contrasts=c('me-other'),levels = design)
fit2=contrasts.fit(fit,cont.matrix)
fit2=eBayes(fit2)
tempOutput = topTable(fit2, coef='me-other', n=Inf)
DEG_limma_voom = na.omit(tempOutput)
head(DEG_limma_voom)
return(DEG_limma_voom) #需要什么就返回什么
}
然后上面原来计算的第一组差异基因就可以用下面简单的代码得到:
group_list=ifelse(df$g==1,'me','other')
deg1=do_limma_RNAseq(exprSet,group_list)
# 再进行一个检验,当然,这里我们也不需要自己去输入基因名
boxplot(dat[rownames(deg1)[1],]~df$g)
同理对第二组进行操作,得到属于第二组的差异基因:
group_list=ifelse(df$g==2,'me','other');table(group_list)
# group_list
# me other
# 300 468
deg2=do_limma_RNAseq(exprSet,group_list)
# 简单做个检查
boxplot(dat[rownames(deg2)[1],]~df$g)
boxplot(dat[rownames(deg2)[2],]~df$g)
接下来就是第三组、第四组:
group_list=ifelse(df$g==3,'me','other');table(group_list)
# group_list
# me other
# 121 647
deg3=do_limma_RNAseq(exprSet,group_list)
group_list=ifelse(df$g==4,'me','other');table(group_list)
# group_list
# me other
# 35 733
deg4=do_limma_RNAseq(exprSet,group_list)
运行完这四个deg就会得到每个组的差异基因,接下来就是挑每个组的top18差异基因(对应原文描述)
如何进行差异基因可视化?
已经确定每个组要挑前18个基因,但是上调下调没确定。如果从图中看,每个组中都是上调的基因,于是我们可以先按照logFC排个序,然后再挑选
deg1=deg1[order(deg1$logFC,decreasing = T),]
deg2=deg2[order(deg2$logFC,decreasing = T),]
deg3=deg3[order(deg3$logFC,decreasing = T),]
deg4=deg4[order(deg4$logFC,decreasing = T),]
# choose gene <=> cg
cg=c(head(rownames(deg1),18),
head(rownames(deg2),18),
head(rownames(deg3),18),
head(rownames(deg4),18)
)
作图开始(一步步进行):
最简单的形式:
library(pheatmap) # 主要就是需要一个表达矩阵 pheatmap(dat[cg,])
加入分组信息
library(pheatmap) ac=data.frame(group=df$g) rownames(ac)=colnames(dat) # 这里不需要列名,显得太乱,而且原文也没有聚类 pheatmap(dat[cg,],show_rownames = T,show_colnames = F, cluster_rows = F,cluster_cols = F, annotation_col = ac)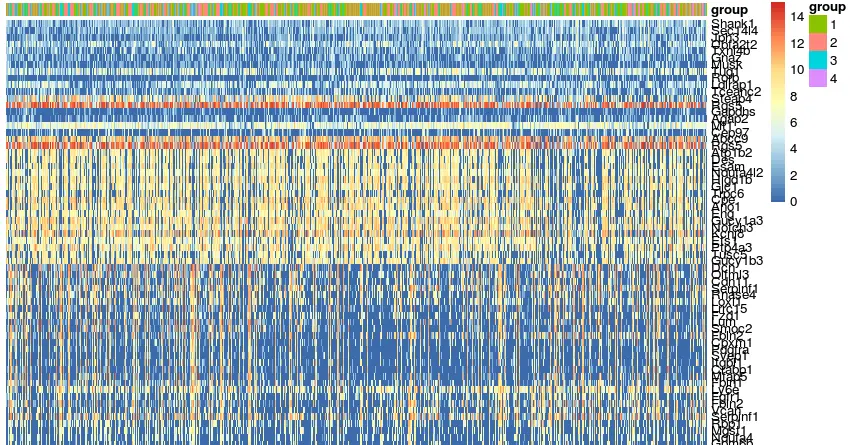
发现的问题是:样本是一个混杂的群体,因此这里分组还是不明了,需要对
df$g重新排序,但是它重新排序了,那么对应的dat[cg,]也要重新排序解决分组排序问题
# 为了下面操作更方便,先将分组和矩阵赋值一个简单的名字 g=df$g mat=dat[cg,] # order()的返回值是对应“排名”的元素所在向量中的位置 mat=mat[,order(g)] ac=data.frame(group=g) rownames(ac)=colnames(dat) pheatmap(mat,show_rownames = T,show_colnames = F, cluster_rows = F,cluster_cols = F, annotation_col = ac)
感觉还不太好,可以进行归一化,先利用函数自带的
scale函数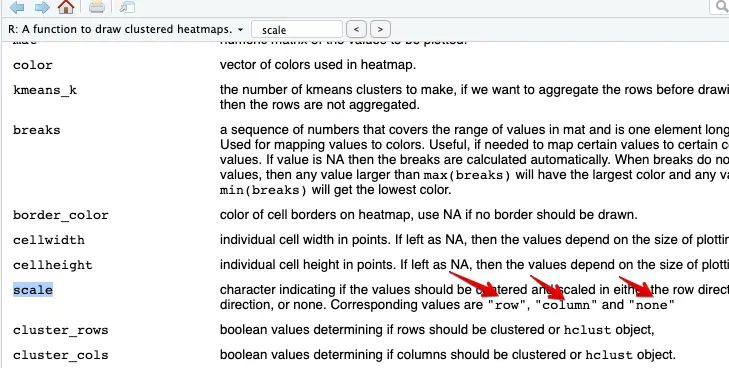
pheatmap(mat,show_rownames = T,show_colnames = F, cluster_rows = F,cluster_cols = F, annotation_col = ac,scale = 'row')但是很丑:
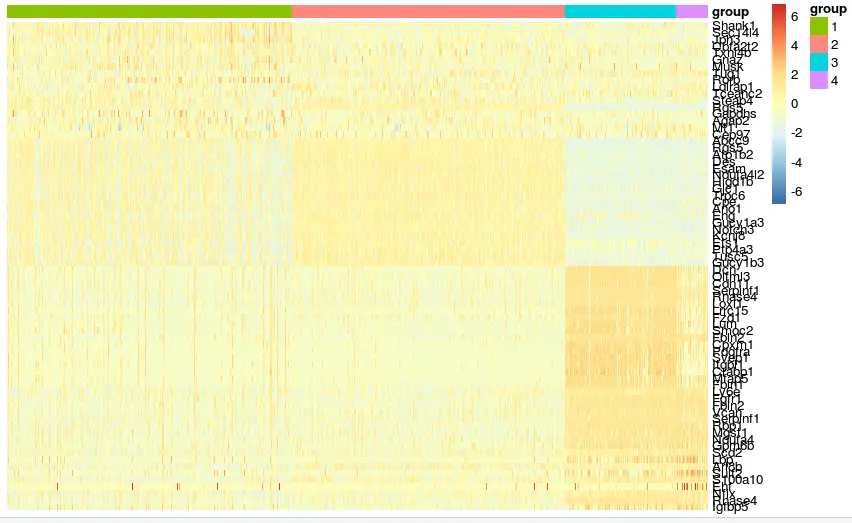
我们自己进行归一化:
n=t(scale(t(mat))) n[n>2]=2 n[n< -2]= -2 # n[1:4,1:4] pheatmap(n,show_rownames = T,show_colnames = F, cluster_rows = F,cluster_cols = F, annotation_col = ac)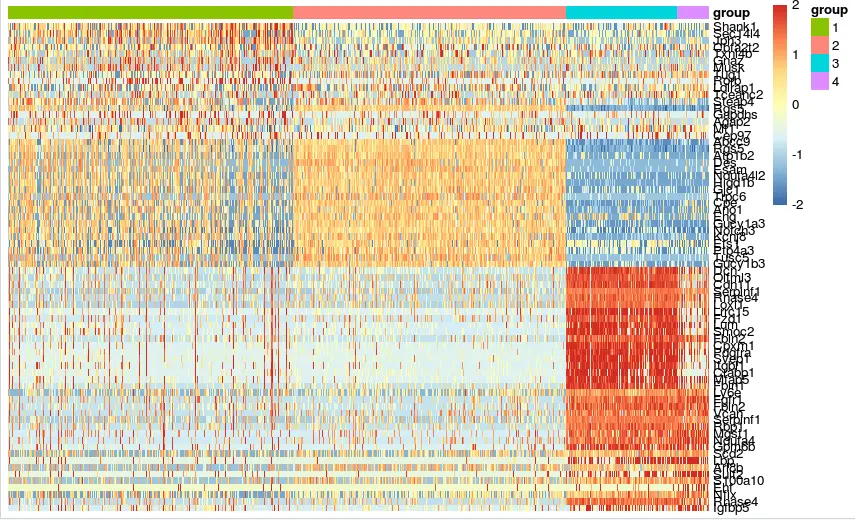
总结差异分析
结果还是没有重现原文的图,可能由于分组和原文不同,因为分组不同就没办法解决异质性的问题,不能将某一撮基因放到一组,也就没办法看到基本上只在这一组内高表达而另外组中低表达的这种情况
对差异分析结果进行操作
首先做个最最简陋的火山图
par(mfrow=c(2,2))
with(deg1,plot( logFC,-log10( adj.P.Val)))
with(deg2,plot( logFC,-log10( adj.P.Val)))
with(deg3,plot( logFC,-log10( adj.P.Val)))
with(deg4,plot( logFC,-log10( adj.P.Val)))
# 下图分别对应1-4组的差异基因
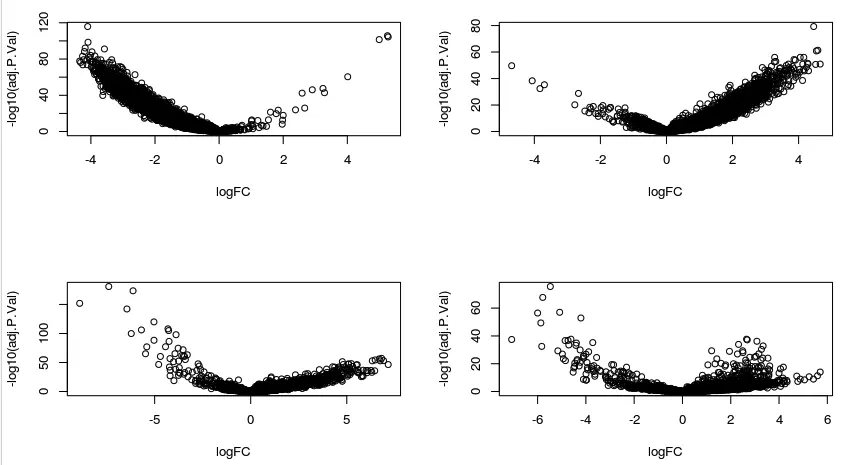
看到这里火山图的形状和我们平常见到的不太一样,这是因为我们得到差异基因的方法存在问题,这里的单细胞数据不单单是原来bulk转录组的 3v3样本这样,每个细胞都是一个样本,而我们只是又将相似的细胞聚在一起当成一个大组,后来我们也是根据大组进行的差异分析(以deg1为例,就相当于312个样本 vs 剩余的456个样本)。另外还是使用的limma包(原文用的
ROTS包),于是产生的差异是可以理解的
接下来过滤差异基因,注释
# 取logFC大于3或者小于-3作为过滤条件
diff1=rownames(deg1[abs(deg1$logFC)>3,])
diff2=rownames(deg2[abs(deg2$logFC)>3,])
diff3=rownames(deg3[abs(deg3$logFC)>3,])
diff4=rownames(deg4[abs(deg4$logFC)>3,])
# 基因ID转换
library(ggplot2)
library(clusterProfiler)
library(org.Mm.eg.db)
df <- bitr(diff1, fromType = "SYMBOL",
toType = c( "ENTREZID"),
OrgDb = org.Mm.eg.db)
kk <- enrichKEGG(gene = df$ENTREZID,
organism = 'mmu',
pvalueCutoff = 0.9,
qvalueCutoff =0.9)
dotplot(kk)
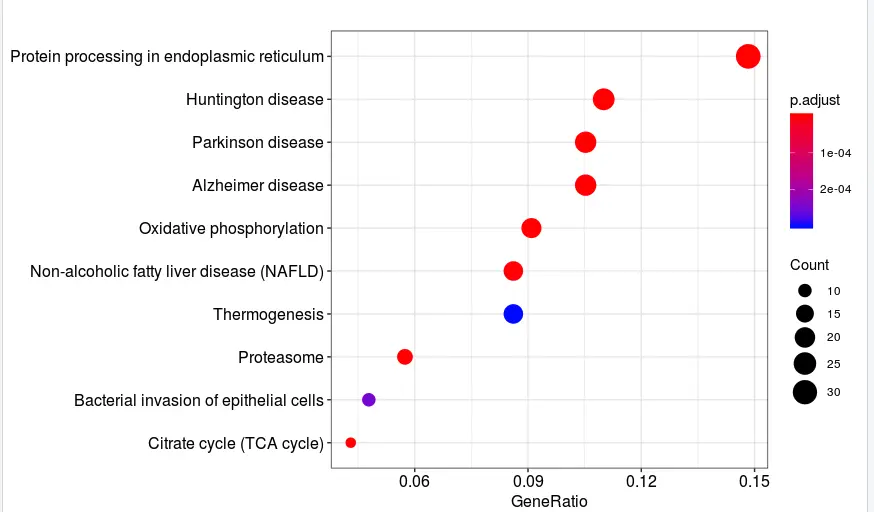
总结
这里只是作为单细胞的入门探索,不是利用真正的单细胞分析算法,算是个过渡阶段吧,接下来会进行真正的单细胞数据之3大R包使用