184-如何根据Entrez ID找基因组坐标?再如果是非模式生物呢?
刘小泽写于2020.5.9 来和我一步步揭开谜团~这一篇对于非模式生物的研究更有帮助哦
前言
昨天晚上帮了朋友一个小忙,需求是:给一些Entrez ID,想找它们的坐标。
如果是模式生物(人、小鼠等),可以直接利用UCSC的BED文件或者GTF/GFF文件寻找,最多进行一下Entrez ID的转换。
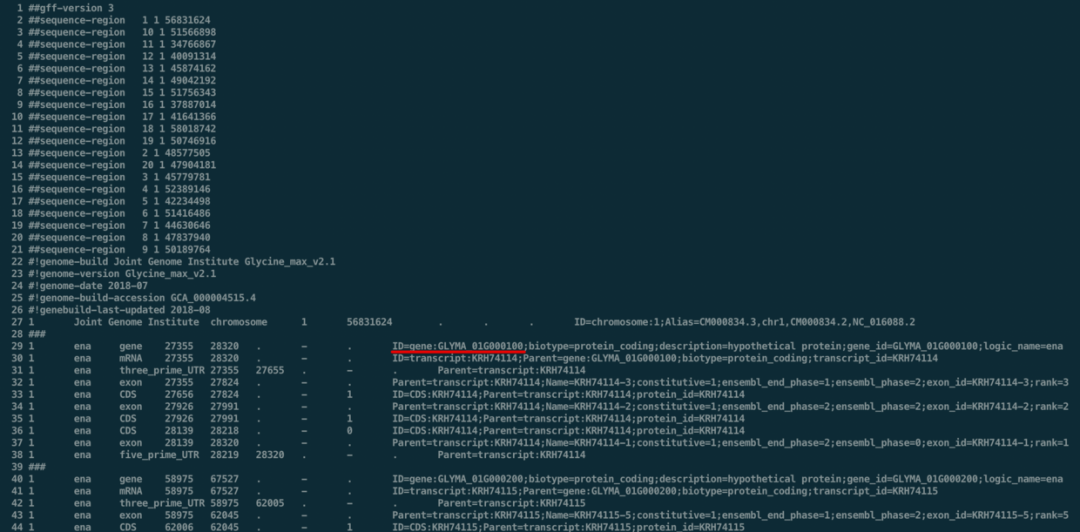
但如果是非模式生物,ID的种类会不太统一。以大豆为例,没有下载到BED文件,但找到了大豆的EnsemblePlant数据库:http://plants.ensembl.org/Glycine_max/Info/Index#,下载到了GFF文件(ftp://ftp.ensemblgenomes.org/pub/plants/release-47/gff3/glycine_max/Glycine_max.Glycine_max_v2.1.47.chr.gff3.gz)。看到这里的ID有点特殊GLYMA_01G000100 ,既不是Entrez,也不是Symbol
先不管了,看看朋友提供的Entrez ID吧:
# 截取了一部分
100785765
100792329
100819570
100789593
100775570
100792423
100793238
100805323
100820568
把第一个100785765放到NCBI Gene中查询,发现了那个奇怪的名称
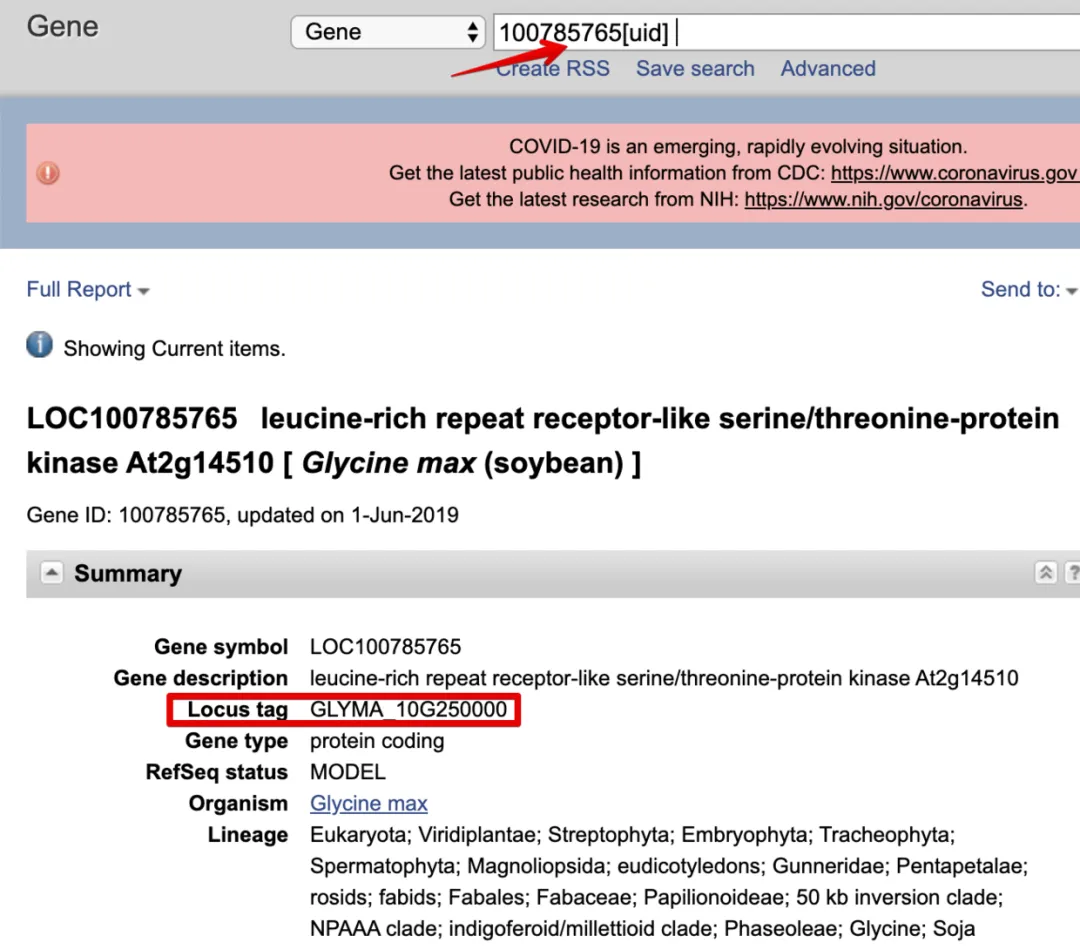
原来这个ID属于Locus tag
其实看到这里,我就心想,用R可能不会成功了,因为物种数据库中不记得有这个类型的ID,但还是想试试
如果用R
重点就是获取大豆的物种注释信息(当然也适用于其他的非模式物种)。
模式物种以及一些常用的物种可以直接获取到Org.db (http://bioconductor.org/packages/release/BiocViews.html#___OrgDb),一共20个
而大豆的学名是:Glycine max ,显然这里没有。于是就要自己构建org.db
library(AnnotationHub)
hub <- AnnotationHub()

query(hub,"Glycine")
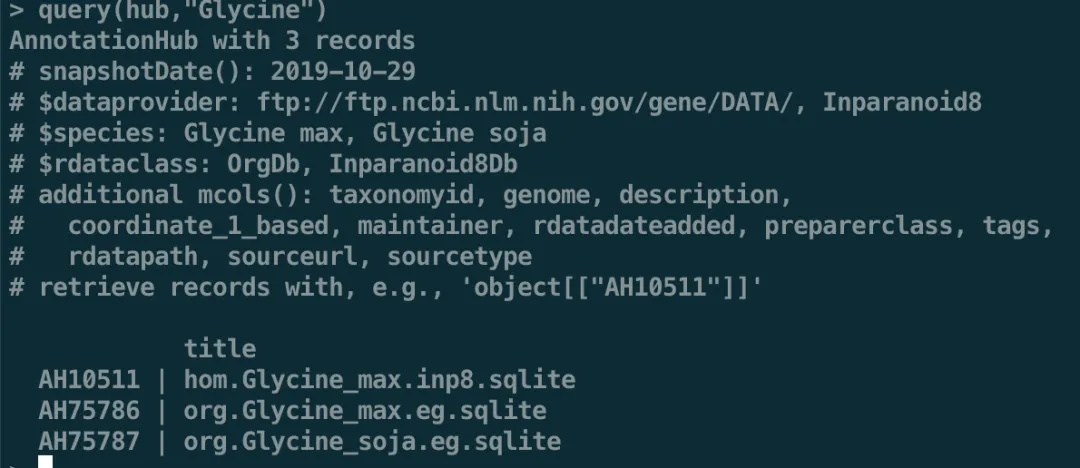
我们可以选第二个:AH75786
Glycine <- hub[["AH75786"]]
> Glycine
OrgDb object:
| DBSCHEMAVERSION: 2.1
| DBSCHEMA: NOSCHEMA_DB
| ORGANISM: Glycine max
| SPECIES: Glycine max
| CENTRALID: GID
| Taxonomy ID: 3847
| Db type: OrgDb
| Supporting package: AnnotationDbi
Please see: help('select') for usage information
看看这个物种数据库包含了什么信息
> keytypes(Glycine)
[1] "ACCNUM" "ALIAS" "ENTREZID" "EVIDENCE" "EVIDENCEALL"
[6] "GENENAME" "GID" "GO" "GOALL" "ONTOLOGY"
[11] "ONTOLOGYALL" "PMID" "REFSEQ" "SYMBOL" "UNIGENE"
然后可以进行ID转换
bitr(genes,fromType = "ENTREZID",toType = "SYMBOL",OrgDb = Glycine)
的确,用R转换比较困难,这里构建的org.db没有Locus Tag,只能另寻它法
偶然间看到了一个网页工具,还不错哦
正在我准备移步DAVID时(不太喜欢用DAVID其实),不死心又尝试搜索了一下
搜关键词:entrez id to Locus tag =》 然后看到:Biostar 中有一个 Question: How To Map Gene Id Or Gene Symbol To Locus:https://www.biostars.org/p/10457/)=》找到了这个工具
网站是:https://biodbnet-abcc.ncifcrf.gov/db/db2dbRes.php
长这样(界面很简洁): 而且支持的ID种类特别多(还有各种芯片ID的对应)

我们把开头的那几个基因贴在这里:

点提交,很快就给出了结果,而且正是想要的!
为了后面的命令行处理,将结果导出为txt【选择Results as Text】
最后就是命令行处理,得到坐标啦
之前那个文件一点都不需要修改(包括第一行是注释信息;并且其中如果有匹配不到而产生的空行也不需要管)
> cat gs.txt
Gene ID Locus Tag
100785765 GLYMA_10G250000
100792329 GLYMA_19G142400
100819570 GLYMA_16G058700
100789593 GLYMA_15G001100
100775570 GLYMA_09G162300
100792423 GLYMA_16G030300
100793238 GLYMA_05G024800
100805323 GLYMA_08G002800
100820568 GLYMA_13G243700
代码逻辑是:
先提取第二列,然后去掉第一行,再去掉空行(如果有的话,使用正则匹配);
之后对每行Locus Tag循环读取,在GFF文件中寻找(而且只要GFF中gene的信息),并将Locus Tag匹配提取出来
cut -f2 gs.txt |sed 1d | sed '/^$/d'| while read i;do grep $i Glycine_max.Glycine_max_v2.1.47.chr.gff3;done | perl -alne '{next unless $F[2] eq "gene"; ;/ID=gene:(.*?);/; print "$F[0]\t$F[3]\t$F[4]\t$1"}'
结果就是:第一列是染色体,第二、三列是起始终止位置,最后是基因名
10 47815076 47819624 GLYMA_10G250000
19 40338311 40341438 GLYMA_19G142400
16 5753646 5756595 GLYMA_16G058700
15 119084 124240 GLYMA_15G001100
9 38644142 38648347 GLYMA_09G162300
16 2892574 2895793 GLYMA_16G030300
5 2170120 2172159 GLYMA_05G024800
8 204552 208237 GLYMA_08G002800
13 35312187 35316963 GLYMA_13G243700