192-基因ID转换升级版探索
刘小泽写于2020.5.28 Ensemble转Symbol其实不是这么简单,问题百出,需要留意
0 今天遇到一个需求,先来看看
有这样的四个文件,每个文件中都有两列Ensembl ID,从命名可以看到是人类的ID

如何辨别Ensembl ID?
- 例如
ENSG00000279928.1中,ENS是固定字符,表示它是属于Ensembl数据库的。默认物种是人,如果是小鼠就要用ENSMUS开头,关于物种代码:http://www.ensembl.org/info/genome/stable_ids/index.html- G表示:这个ID指向一个基因;E指向Exon;FM指向 protein family;GT指向gene tree;P指向protein;R指向regulary feature;T指向transcript
- 后面11位数字部分如
00000279928表示基因真正的编号- 小数点后不一定每个都有,表示这个ID在数据库中做了几次变更,比如
.1做了1次变更,在分析时需要去除至于怎么去掉小数点后面的信息也很简单,之前花花介绍过( 去掉ensembl id的version信息)
然后每一行表示:一个基因(第一列)与另一个基因(第二列)编码的蛋白存在互作关系
于是可以看到很多这样的情况:一个基因(例如ENSG00000000005)与其他十几个基因都有关系。我们需要保证转换后的顺序和原来的顺序一一对应
1 先处理一个文件的转换
既然有两列,那思路就是:先对第一列的id进行转换,然后按照原来的顺序添加到新的一列,即第三列;再对第二列的id进行转换,然后按照原来的顺序添加到新的一列,即第四列
1.1 首先读取数据看看
rm(list=ls())
options(stringsAsFactors = F)
library(tidyverse)
library(org.Hs.eg.db)
library(clusterProfiler)
library(stringr)
df=read.csv('test1.tsv',sep="\t",header = F)
> dim(df)
[1] 64006 2
# 这里的6万多行不是真实的6万个基因,因为存在大量的重复ID情况
> length(unique(df$V1))
[1] 6919 # 第一列实际有6919个基因
> length(unique(df$V2))
[1] 7162
1.2 然后对第一列进行转换
# drop参数:drop NA or not
conv=bitr(df$V1,fromType = "ENSEMBL",
toType = "SYMBOL",OrgDb = org.Hs.eg.db, drop = F)
> dim(conv)
[1] 6980 2
# 看到相比原来的6919个基因,少了一些,就要想到Ensemble ID对应Symbol时,存在一对多的关系,即一个Ensemble 可能对应多个Symbol,检查一下
conv[which(duplicated(conv$ENSEMBL)),]
# 如果一个ID出现了2次或者多次,那就会返回这些重复的位置,再取出来看看
1.3 特殊情况一:存在一对多
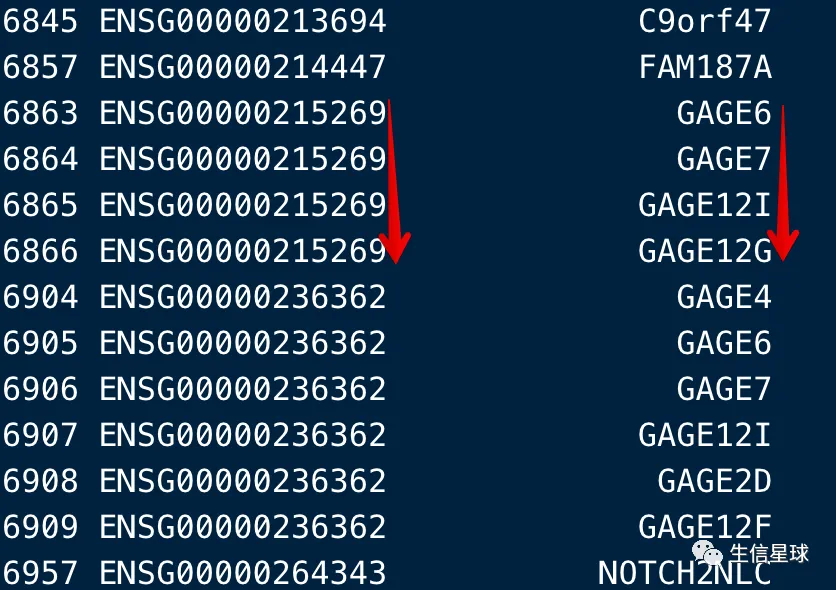
看到这4个ENSG00000215269,实际上总共出现了5次,在6862这个位置是第一次出现,之后的6863-6866这几个symbol就被认定为了重复
> conv[6862,]
ENSEMBL SYMBOL
6862 ENSG00000215269 GAGE4
既然存在一对多,那么就应该保留唯一的一个symbol
一般为了代码的便利性,会选择第一个,但第一个对不对呢?这里举两个例子:
1.3.1 第一个例子:ENSG00000215269
library("AnnotationDbi")
# 第一个例子:ENSG00000215269
geneSymbols <- mapIds(org.Hs.eg.db, keys='ENSG00000215269', column="SYMBOL", keytype="ENSEMBL", multiVals="CharacterList")
> unlist(geneSymbols)
ENSG00000215269 ENSG00000215269 ENSG00000215269 ENSG00000215269 ENSG00000215269
"GAGE4" "GAGE6" "GAGE7" "GAGE12I" "GAGE12G"
# 第一个是GAGE4,最后一个是GAGE12G
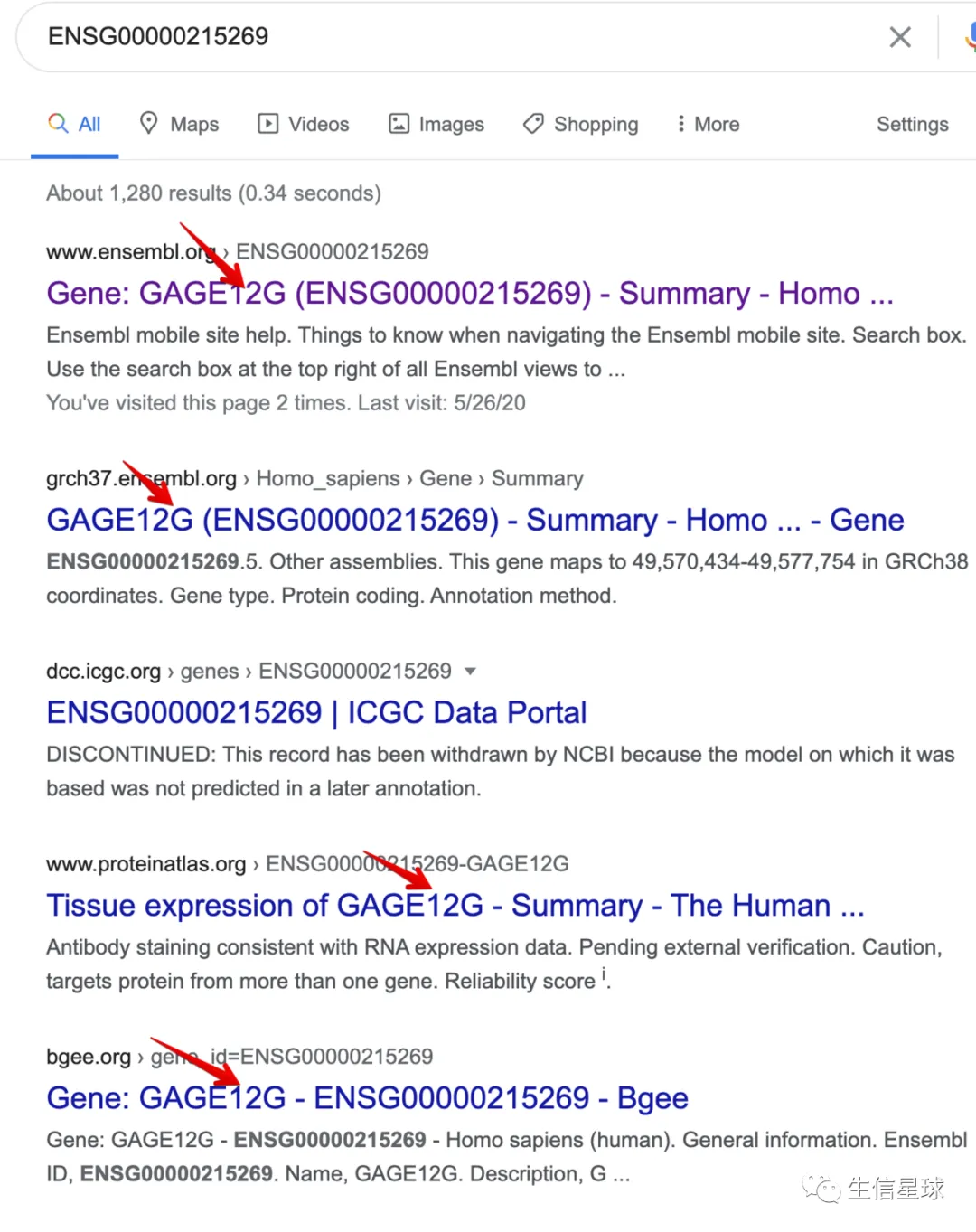
但是当把这个Ensemble ID去搜索,会发现并不是第一个,而应该是最后一个GAGE12G:
再次检索一下第一个GAGE4基因:
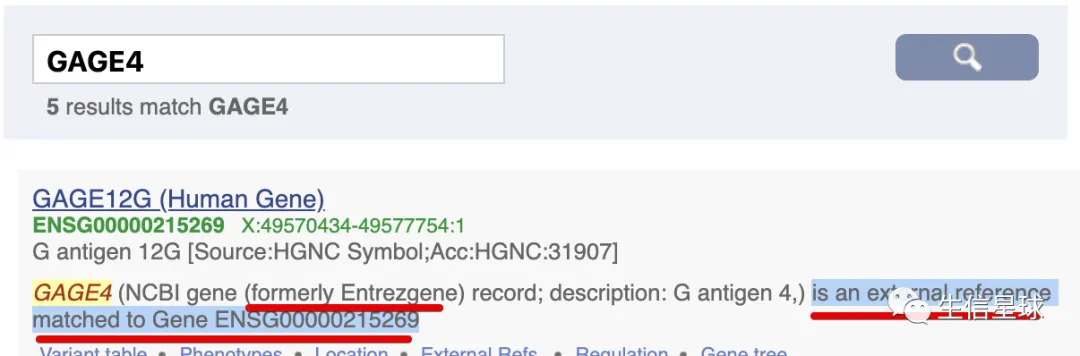
对比一下二者:
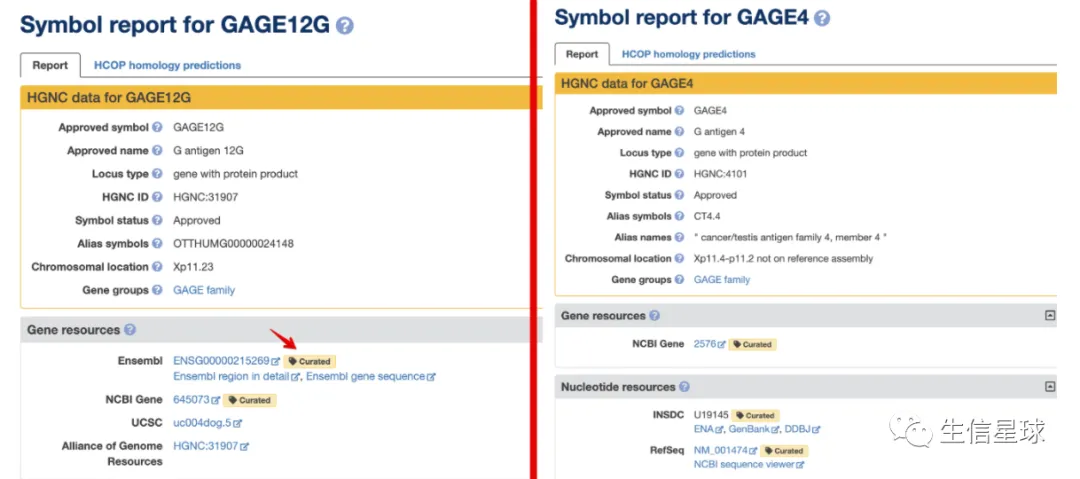
这时会想,那我不用第一个了,用最后一个呗。事情依然没有这么简单:
1.3.2 第二个例子:ENSG00000063587
可以看到这个ENSG00000063587基因存在两个symbol:ZNF275和LOC105373378
> conv[389:392,]
ENSEMBL SYMBOL
389 ENSG00000063515 GSC2
390 ENSG00000063587 ZNF275
391 ENSG00000063587 LOC105373378
392 ENSG00000063854 HAGH
搜索一下就知道,这第一个symbol:ZNF275 是可靠的
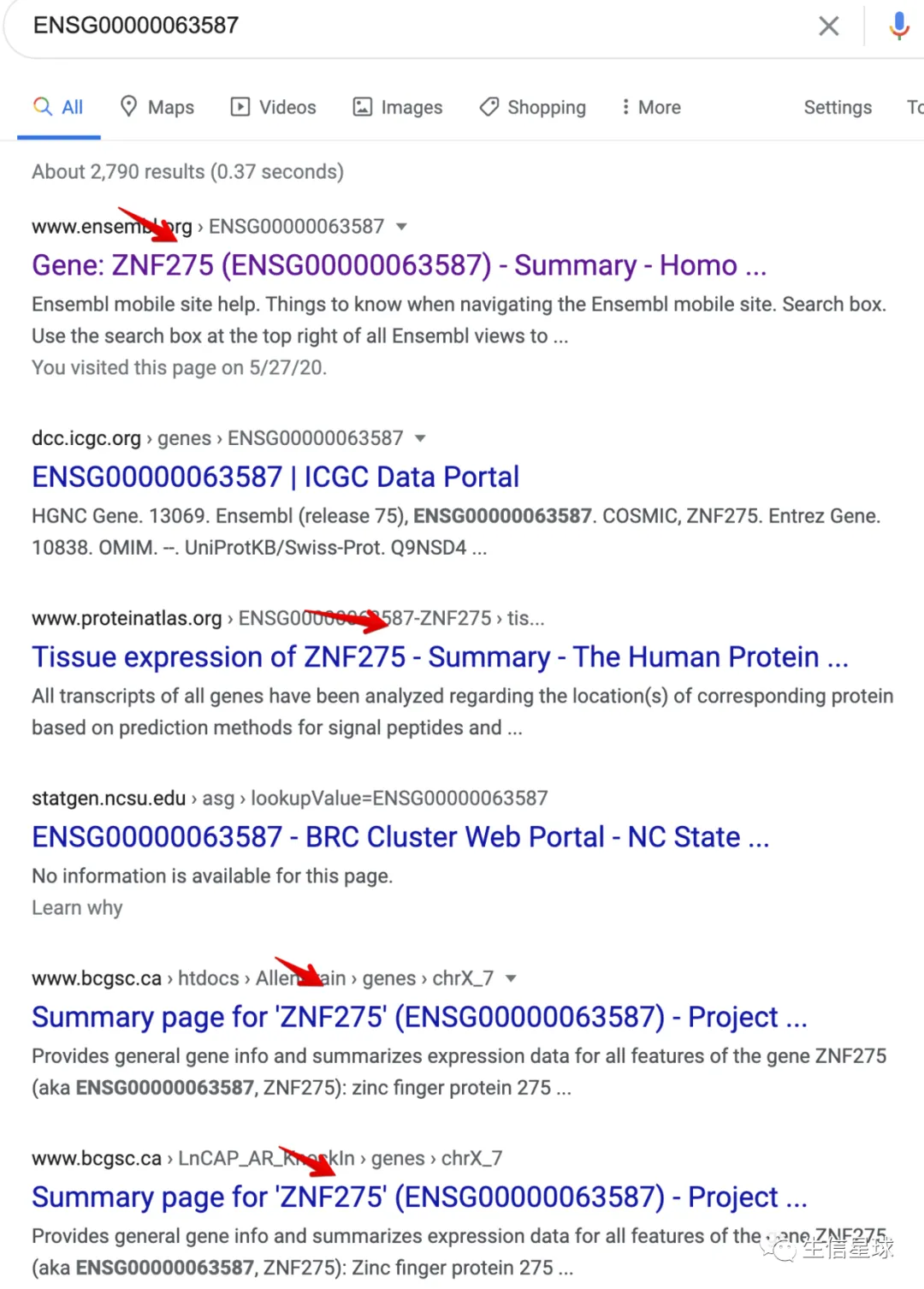
好,用第一个也不对,用最后一个也不对,那应该怎么办呢? 这时要考虑换一下注释数据库
ID转换方法不止一种,详见之前我写的: gene的symbol与entrez ID并不是绝对的一一对应的
尤其是针对Ensemble ID时,自家的转换能获得更多的结果
library("biomaRt")
# 用useMart函数链接到人类的数据库
ensembl <- useMart("ensembl", dataset = "hsapiens_gene_ensembl")
# 除以以外还有很多:使用 listDatasets(ensembl) 查看
attributes <- listAttributes(ensembl)
View(attributes)
value <- df$V1
attr <- c("ensembl_gene_id","hgnc_symbol")
ids <- getBM(attributes = attr,
filters = "ensembl_gene_id",
values = value,
mart = ensembl)
# 如果有匹配不上的,还是用原来的Ensembl ID
ids$hgnc_symbol[ids$hgnc_symbol == ""] <- ids$ensembl_gene_id[ids$hgnc_symbol == ""]
ids
1.4【补充】特殊情况二:存在多对一
这一小部分只是为了文章结构的完整性,这里的数据没有体现 那么只存在一对多吗?并不是!还有多个Ensemble 对应一个symbol
看:https://www.researchgate.net/post/How_to_deal_with_multiple_ensemble_IDs_mapping_to_one_gene_symbol_in_a_RNA-Seq_dataset

但其实看一下Ensemble ID在染色体上的位置就能知道:
只有下图中加粗的那个ID才位于染色体的主体(chr19),其他的都来自haplotypic regions
名词解释: Haplotyptic regions are defined by the Genome Reference Consortium (GRC) and are visible on all their genome assemblies for human, mouse, and zebrafish. They can also be called ‘alternate locus’, ‘partial chromosomes’, and ‘alternate alleles’ or ‘assembly exceptions’ These regions can have small sequence differences, contain different gene structures or different genes entirely, or contain genes in a different order compared to the reference genome assembly.
2 下面对比bitr和biomart
下面的conv是bitr的结果,ids2是biomart的结果,df是原来的ID数据框
conv2=conv[!duplicated(conv$ENSEMBL),] # 简单去重,只保留第一个
df2=left_join(df,conv2,by=c("V1"="ENSEMBL")) # bitr结果
df3=left_join(df,ids2,by=c("V1"="ensembl_gene_id")) # biomart结果
> dim(df);dim(df2);dim(df3)
[1] 64006 2
[1] 64006 3
[1] 64006 3
# 上面的第一个例子:ENSG00000215269
> df2[df2$V1=='ENSG00000215269',]
V1 V2 SYMBOL
63685 ENSG00000215269 ENSG00000283632 GAGE4
> df3[df3$V1=='ENSG00000215269',]
V1 V2 hgnc_symbol
63685 ENSG00000215269 ENSG00000283632 GAGE12G
看看df2中一些NA的情况:例如ENSG00000064489在bitr中没有,但在biomart中有
https://asia.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000064489;r=19:19145569-19192158
df2[df2$V1=='ENSG00000064489',]
df3[df3$V1=='ENSG00000064489',]
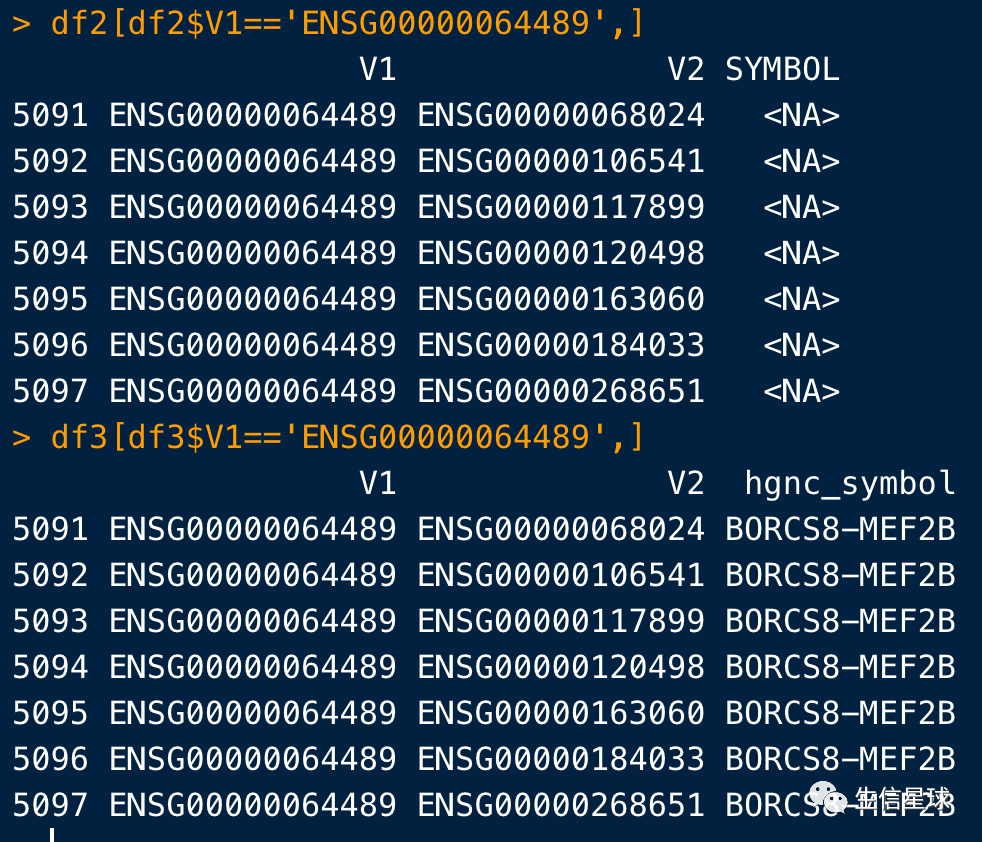
**但能说biomart就一定比bitr好吗?**显然不能这么说,例如ENSG00000036549就在bitr结果中有,而在biomart结果没有
# 再看一个例子:
df2[df2$V1=='ENSG00000036549',]
df3[df3$V1=='ENSG00000036549',]
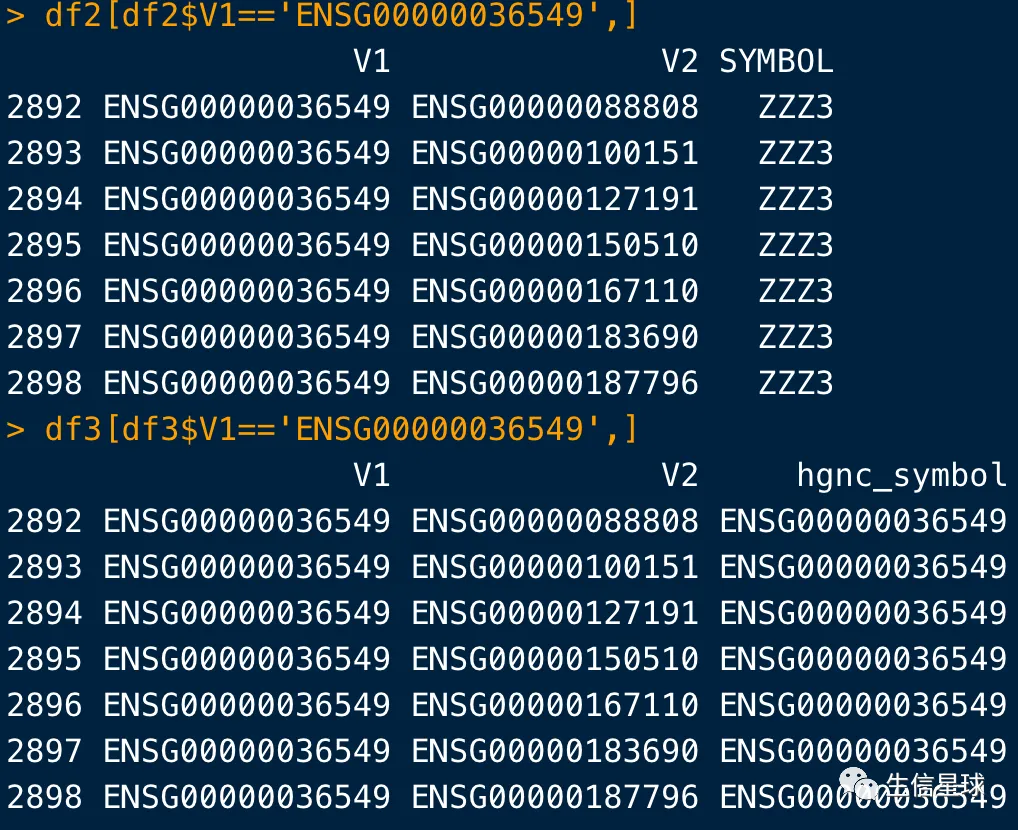
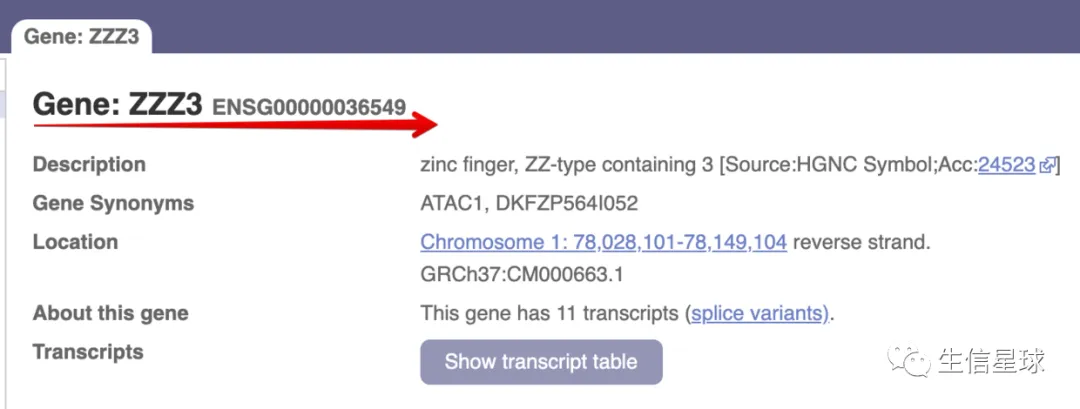
并且搜索验证一下,ENSG00000036549 还真的有这个Symbol ID对应
不过这里是GRCh37 (hg19)基因组版本
http://grch37.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000036549;r=1:78028101-78149104
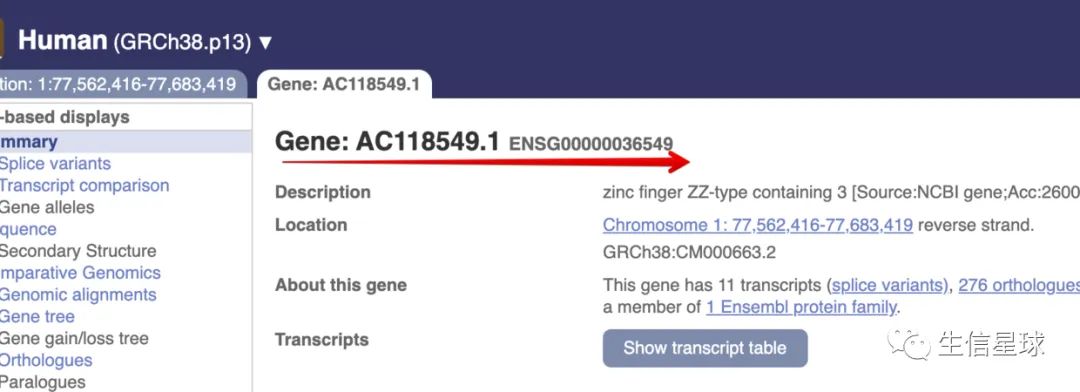
如果是GRCh38(hg38)的话,结果就是:
http://asia.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000036549;r=1:77562416-77683419
3 代码整合版
上面的内容作为探索过程,理解做了什么就好
df=read.csv(files[i],sep="\t",header = F)
## 用biomart转换id
# 用useMart函数链接到人类的数据库
ensembl <- useMart("ensembl", dataset = "hsapiens_gene_ensembl")
# 除以以外还有很多:使用 listDatasets(ensembl) 查看
# attributes <- listAttributes(ensembl)
# View(attributes)
## 对第一列操作
value <- df$V1
attr <- c("ensembl_gene_id","hgnc_symbol")
conv <- getBM(attributes = attr,
filters = "ensembl_gene_id",
values = value,
mart = ensembl)
# 如果有匹配不上的,还是用原来的Ensembl ID
if(sum(conv$hgnc_symbol == "") != 0){
conv$hgnc_symbol[conv$hgnc_symbol == ""] <- conv$ensembl_gene_id[conv$hgnc_symbol == ""]
}
# 存在Ensemble与Symbol一对多的情况,去重
conv2=conv[!duplicated(conv$ensembl_gene_id),]
# dim(conv);sum(duplicated(conv$ensembl_gene_id));dim(conv2)
# 没有对应存为NA
df2=left_join(df,conv2,by=c("V1"="ensembl_gene_id"))
## 对第二列操作
value <- df$V2
attr <- c("ensembl_gene_id","hgnc_symbol")
conv3 <- getBM(attributes = attr,
filters = "ensembl_gene_id",
values = value,
mart = ensembl)
# 如果有匹配不上的,还是用原来的Ensembl ID
if(sum(conv3$hgnc_symbol == "") != 0){
conv3$hgnc_symbol[conv3$hgnc_symbol == ""] <- conv3$ensembl_gene_id[conv3$hgnc_symbol == ""]
}
conv4=conv3[!duplicated(conv3$ensembl_gene_id),]
# dim(conv3);sum(duplicated(conv3$ensembl_gene_id));dim(conv4)
df3=left_join(df2,conv4,by=c("V2"="ensembl_gene_id"))
df3[,3][which(is.na(df3[,3]))]=df3[,1][which(is.na(df3[,3]))]
df3[,4][which(is.na(df3[,4]))]=df3[,2][which(is.na(df3[,4]))]
4 为何不强强联合呢?
既然看到有少部分在bitr有注释,但biomart没有 那么,在biomart的基础上加上bitr的补充结果,岂不是更好?
# 就是看看hgnc_symbol这一列中显示ENSG0*****的基因在bitr中能不能转换成功,能的话就取代原来的结果
## 整合bitr
# 先得到org.Hs.eg.db中的所有Ensemble ID
org_ensembl=toTable(org.Hs.egENSEMBL)[,2]
# 对第一列操作
for(i in 1:length(df3$hgnc_symbol.x)){
name=df3$hgnc_symbol.x[i]
# 判断这里的name是不是在org.Hs.eg.db中(org_ensembl)
if(str_detect(name,"ENSG0") & name %in% org_ensembl){
df3$hgnc_symbol.x[i]=bitr(name,fromType = "ENSEMBL",
toType = "SYMBOL",
OrgDb = org.Hs.eg.db, drop = F)[,2]
}
}
# 对第二列操作
for(i in 1:length(df3$hgnc_symbol.y)){
# i=1
name2=df3$hgnc_symbol.y[i]
if(str_detect(name,"ENSG0") & name2 %in% org_ensembl ){
df3$hgnc_symbol.y[i]=bitr(name,fromType = "ENSEMBL",
toType = "SYMBOL",
OrgDb = org.Hs.eg.db, drop = F)[,2]
}
}
5 小结
当转换Ensemble时,最好还是用自家的biomart,可以注释到更多的Ensemble ID,准确性也会更好一些,但速度可能会稍微慢一点点;
如果基因ID数量不多,只是想快速看看转换后的ID,用bitr 也是足够的
最后,用了一个上午重新探索了一下ID转换,最后将bitr和biomart合二为一,增加了ID转换的准确性和丰富性,并且可以批处理多个文件,还增加了一些小修饰(比如每个文件转换的时间计算、转换结果输出前进一步检查,保证转换前后的结果每行的Ensemble ID一致)