004-测序的世界
刘小泽写于18.6.12
测序的世界很奇妙,不同的数据处理可能得出不同的结论,入门生信首先要做的就是了解你的数据
还等什么?跟我一起来探索吧~
主要从以下四部分详细展开:
测序原理– 数据产生 – 数据初步分析–数据过滤
测序原理:
早期如何测序呢?
如果给你这么一串珠子,让你统计绿色,白色,红色,其他这四种类型的数量,那你是不是要从某个位置一个一个数?没错,你已经开始对这串珠子进行了肉眼测序。

其实对于DNA测序,思路也是这样。
早在1954年,Whitfeld等就提出了测定多聚核糖核苷酸链的降解法,该方法利用磷酸单酯酶的脱磷酸作用和高碘酸盐的氧化作用从链末端逐一分离寡核糖核苷酸并测定其种类。目的就是想通过这种一个一个“数”的方法来得到DNA的碱基顺序。
这里再补充一个小知识,DNA有多大?它的直径也就2nm,两个核苷酸之间的小沟0.34nm。肉眼观察肯定不行,那么显微镜呢?光学显微镜可见光的波长约为400~700nm,单单一个直径就看不到,更别说看更小的分子了DNA对于它来说根本不可见。要想用普通显微镜观察不可能,更强大的电子显微镜呢?别忘了,DNA分子并不是平躺着让你去观察,他有四级结构,不断扭曲、折叠,简单的二维图片是很难区分开来的,能看到也仅仅只能看到一小部分。就像上面的这张珠子图,拍一张照片,就能准确区分被压着的珠子和它上面的珠子顺序吗?不能够!
人类基因组共有31.6亿个DNA碱基对,23对染色体,测一条染色体就需要测1-2亿个碱基对,像这种数数的办法肯定行不通,太耗费人力。那么有没有什么办法可以实现半自动化呢?
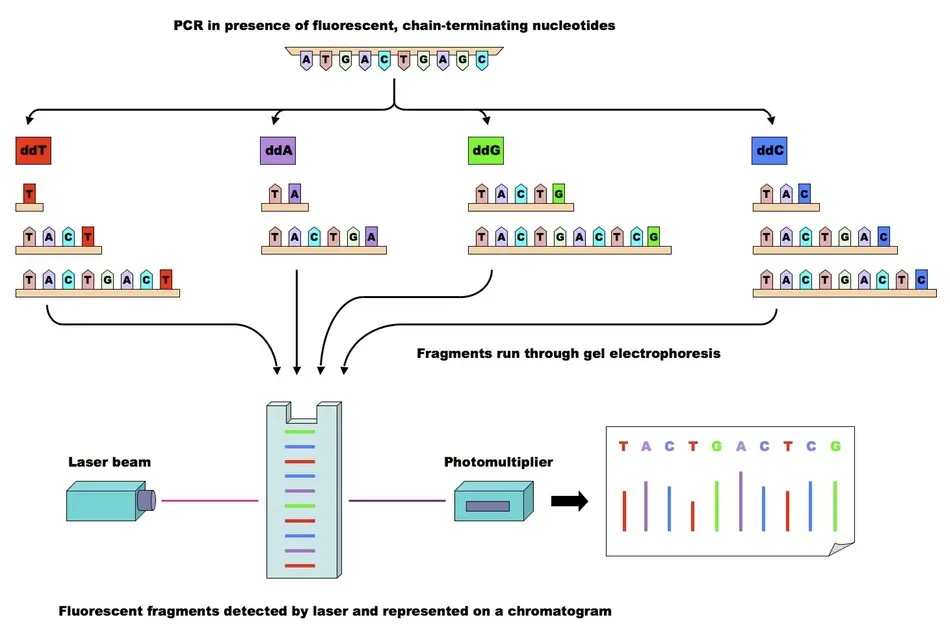
视觉不行,另辟蹊径,听过“盲人摸象”的故事吧,70年代的Sanger发明了“双脱氧终止反应法”,他就是利用了双脱氧核苷酸 ddNTP去摸索DNA分子。例如,一条序列5‘ –> 3’ 实际为TGACTTCG但我们之前不知道,这样操作:
操作过程可能会有些无聊,但是不难懂,希望你能理解其中的意思,因为后来测序都是受此启发,这是鼻祖
设置四个反应体系1-4,分别加入引物、DNA聚合酶、四种dNTP、一定比例的ddNTP(带有放射性标记) 例如1中是ddATP,它就负责测定T碱基的位置;依次2是ddCTP,3是ddTTP, 4是ddGTP
假如扩增过程中ddATP遇到了T位点,就结合并终止(因为ddNTP的2‘和3’都没有羟基),那么其他的dNTP就无法结合。也许你会问,那么每次如果是这四种ddNTP占了先机先结合了,其他的都无法再结合,结果不就是一种可能,只能确定四个位点? 并不是这样: 大批量的核苷酸结合本就是一个随机问题,不一定每次都是ddNTP结合,有可能是dNTP结合了好几个然后ddNTP才回过神,结合完再宣告一个聚合过程结束。
想象ddNTP是VIP会员,dNTP为普通用户,只要ddNTP不结合(不占用绿色通道),普通的dNTP就有机会。
因此,在这反反复复的结合过程中,就会把所有的位点都结合,这其中有的是dNTP结合,有的是ddNTP结合,但是,ddNTP确实结合了所有位点,只是位点结合次数多少的问题。 说的简单一点就是,一段时间内大量的ddNTP会结合完所有测序位点。
Q: 最后怎么看结果?这里很有意思,一个位点可能会被大量的dNTP和ddNTP结合,怎么去除dNTP的干扰呢?
A: 其实也不用刻意去除,最后利用凝胶电泳和放射自显影只能看到带有荧光标记的ddNTP,他们的排列顺序先利用电泳条带前后关系确定下,再用A-T, T-A, C-G, G-C关系反转一下,就能知道我们的测序序列
附上一张图,帮助理解
有了早期的第一次测序成功,才有了后来1983年的Kary Mullis 发明PCR测序仪,利用PCR才有了我们更加效率的NGS(二代测序)。进步的是方法,不变的是基本理念。
二代测序:
一代测序的原理我们搞清楚了,没有PCR,利用聚合酶延伸链。这就有一个问题,酶的活性会下降,所以一代最长能测1000bp,再多了就要重头开始一遍导致成本高;另外它一次只测一条,也就是所谓的通量低。但是呢,他的确有值得借鉴的地方,就是准确度很高,99.999%。人类基因组计划HGP,1990年-2003年完成,利用的就是改进的Sanger测序法
后来技术不断改进,Roche公司的454技术、illumina公司的Solexa/Hiseq技术和ABI公司的SOLID技术标志第二代测序技术诞生。其中Roche公司的454测序系统是第二代测序技术中第一个商业化运营的测序平台。
其中Illumina市场规模占到75%以上,主打Hiseq,下面👇就主要介绍他的PE(Pair End双端)测序原理:
必备名词:
flowcell: 测序反应的载体/容器,1个flowcell有8个lane
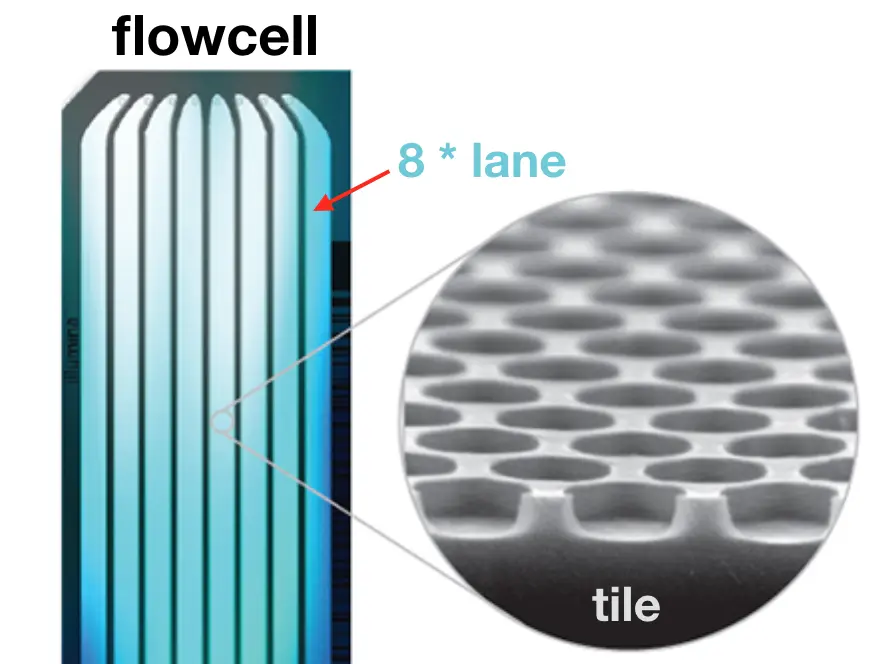
lane: 测序反应的平行泳道,试剂添加、洗脱等过程的发生位置
tile: 每次荧光扫描的位置,肉眼是看不到的
双端测序: 可能序列比较长有四五百bp,两边各测120-150bp
junction: 双端测序中间一些没有测到的区域
**构造:**一个lane包含两列(swath),每一列有60个tile,每个tile会种下不同的cluster,每个tile在一次循环中会拍照4次(每个碱基一次)
边合成变测序(sequence by synthesis, SBS)~合成
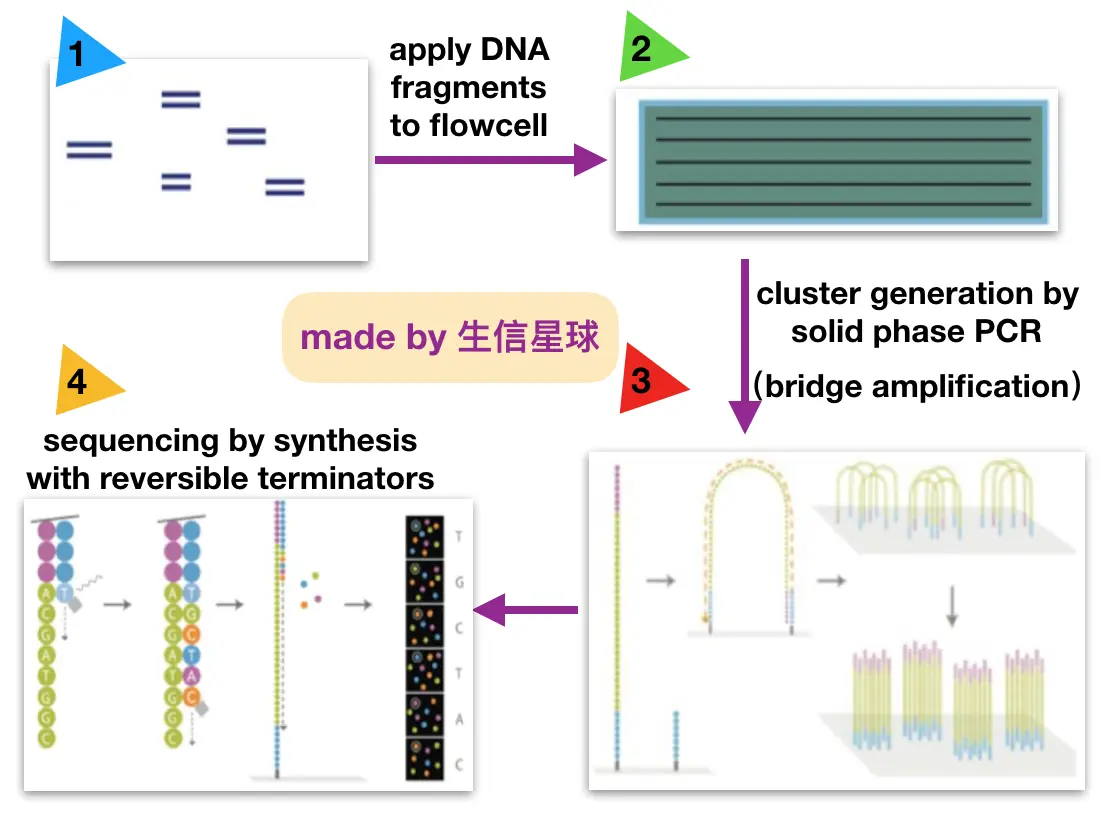
<图一>第一步: 构建DNA文库
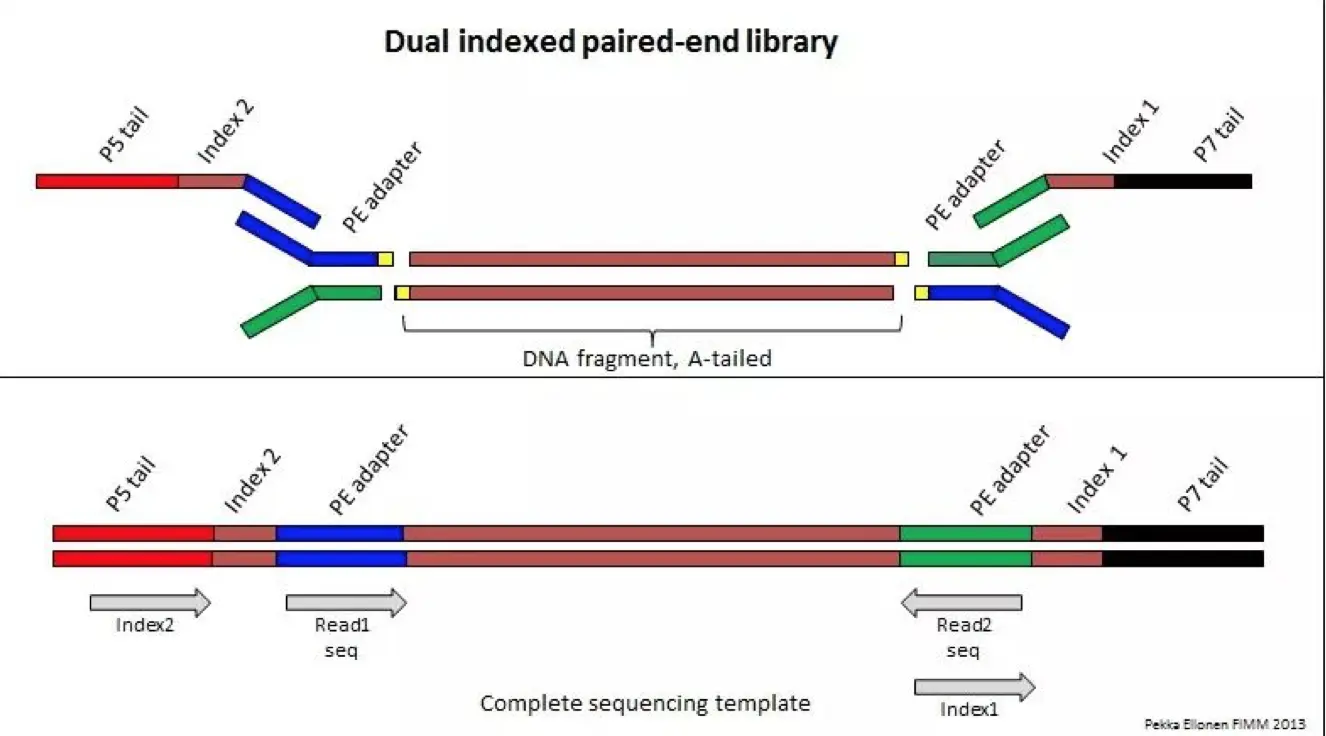
超声波将DNA分子打断成300-800bp长序列片段(人类基因组打成300-500bp),用酶补平为平末端,然后3‘端加一个A碱基(因为接头的3‘端有一个突出的T),再在两端加上互补配对的adapter,再通过PCR扩增达到一定浓度,构成单链DNA文库。【接头设计很巧妙,两个作用:1. 实现桥式扩增,高效;2. 可以实现双端测序】
**好奇,接头怎么加上去的呢?**来源https://www.fimm.fi/en/services/technology-centre/sequencing/next-generation-sequencing/dna-library-preparation
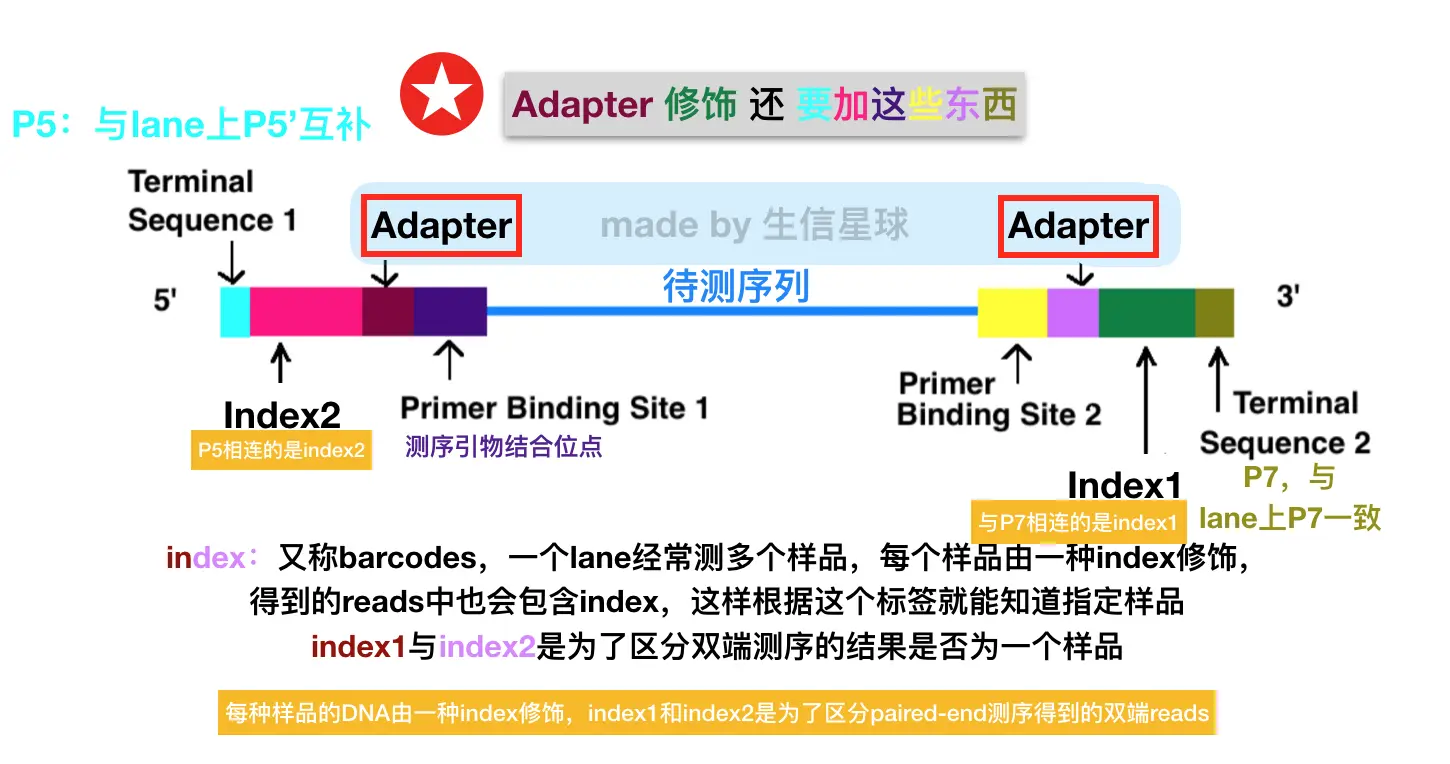
另外,利用低循环扩增技术在接头处进行修饰加上一些周边。
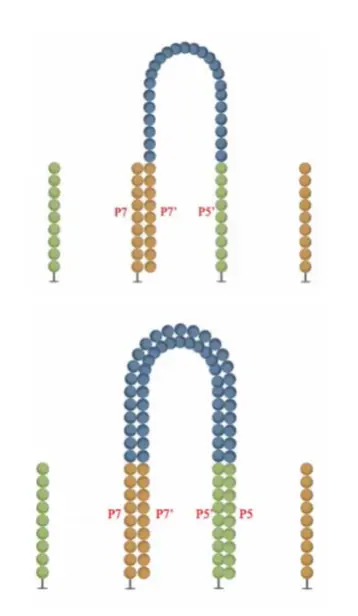
<图二>第二步: 上样 【重点搞清楚lane上有哪两种接头,待测序列含有哪两种接头,这很重要!】
- flowcell是用于吸附流动DNA片段的槽道,测序就在此进行。上面构建好的文库中的待测序列事先配置好一定的浓度,经过这里的时候,会在特异的化学试剂作用下,强力随机地附着在lane上,与上面的短序列配对。上样的结果就是lane吸附住了冲过来的DNA,并且可以在表面进行桥式PCR扩增。
- 【⚠️这里有三点要注意:1. lane与lane之间一般不会相互影响,也就是说一般不会出现lane1固定的DNA又与lane2结合。2. lane上随机分布两种接头,p5‘(与P5互补,蓝色),P7(与P7’互补,紫色)。待测序列自带了p5接头和p7接头; 3.序列只能一开始是利用p5接头互补,因为p7接头和lane是一样的嘛】
<图三>第三步:桥式PCR
第一轮扩增模版:flowcell表面固定的序列 –> 模版链
第一轮结果:序列补成双链。
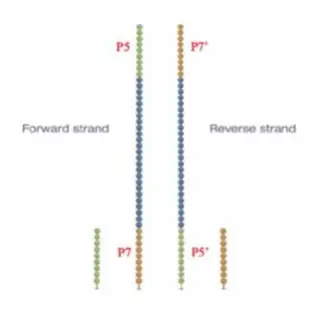
**一个很重要的概念:**我们要测序的是模版链p5 - p7,开始它与lane接头配对产生了互补链,后来强碱试剂作用下两条互补链被分开,由于模版链没有附着在lane上,模版链被冲走,但是互补链依然稳稳固定在lane上。接下来就要对互补链p5‘- p7‘ 进行操作~
去杂:加入NaOH强碱性溶液使双链DNA变性,互补链由于和lane上短序列强力连接固定住了;模板链失去了双链氢键连接,好似悬空,它会被洗脱
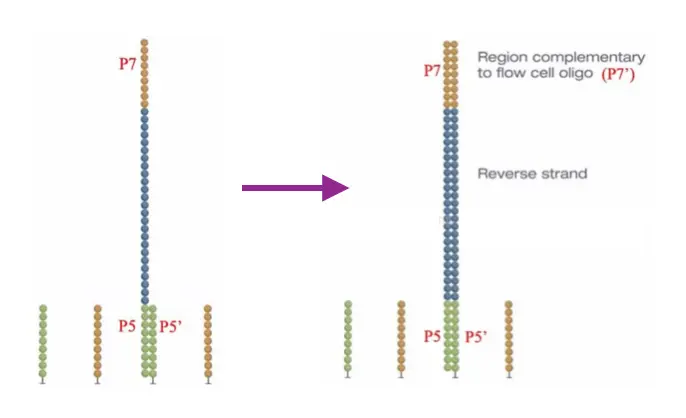
桥式形成: 加入缓冲溶液,互补链的p7‘和lane上的p7互补(但还是一个lane中的)就像下图这样(摘自illumina官网)目的是快速扩增lane p7接头连接的链,也就是下图中的Forward Strand,它和我们的模版链是一致的。我们后来测序只用这一半
- 桥式PCR: PCR弯成桥状,一轮桥式扩增一倍
- 循环: 大约35个循环后,最终每个DNA片段都将在各自的位置上集中成束,称为cluster,这是一群完全相同的序列。目的在于实现放大单一碱基的信号强度,满足后期测序需求
- 解链: 桥式PCR完成后,形成了很多的桥形的互补双链,再次强碱解链。这一次不再进行复制,而是利用一种酶–甲酰胺基嘧啶糖苷酶(Fpg)选择性的切掉lane 上p5‘ 连接的链,只留下了与lane p7连接的链即Forward Strand
<图四>第四步:测序
如何确定上面👆形成的cluster的碱基排序顺序呢?illumina采取了“一次加一个荧光碱基,用完失效”的办法。官网给出的解释如下图:【有没有感觉和Sanger的方法很像?illumina的测序就是在Sanger基础上加上了桥式PCR,能克服Sange低通量的缺点】
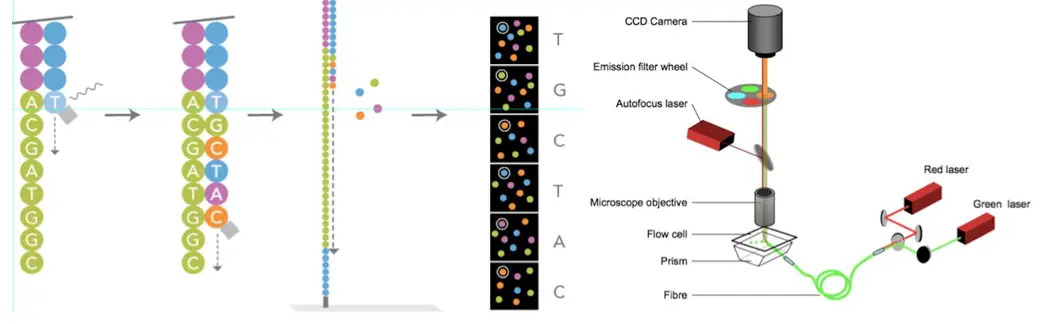
边合成变测序~测序
一轮测序是这样完成的:
双端测序之Forward Strand
先是primer结合到靠近p5的sequencing primer binding site1上,再加入特殊的dNTP【它的3‘ 羟基被叠氮基团替代,因此每次只能添加一个dNTP;还含有荧光基团,能激发不同颜色】;
在dNTP被添加到合成链上后,所有未使用的游离dNTP和DNA聚合酶会被洗脱掉;
再加入激发荧光缓冲液,用激光激发荧光信号,光学设备记录荧光信号的记录,计算机将光学信号转化为测序碱基
这一个循环就能测定flowcell上成千上万的cluster,这就实现了高通量
再向下一轮:
再加入化学试剂淬灭荧光信号并使dNTP 3’ 叠氮基团变成羟基,这样能继续向下进行再加一个,并且保证这个不再发出荧光。如此重复直至所有链的碱基序列被检测出。得到了Forward Strand序列
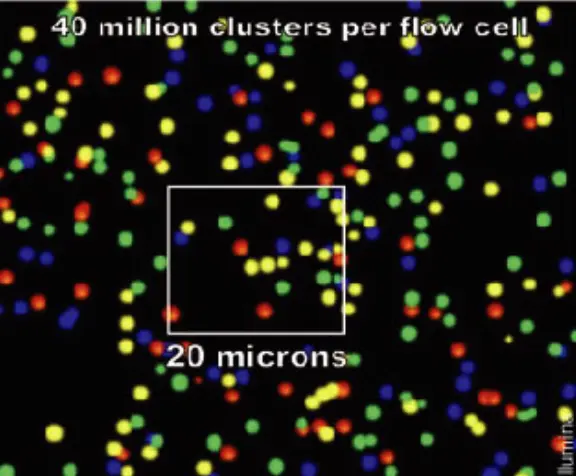
因为一个cluster的序列是一样的,所以理论上cluster的荧光颜色应该一致,下面是来自网站http://tucf-genomics.tufts.edu/home/faq的扫描图片。本来仪器得出的是黑白的,颜色是后来计算机计算分析后加上去的
Index测序: 上面的循环结束后,read product被冲掉,index1 primer和链上的index1 互补配对,进行index1的检测。测完后,洗脱产物,得到index1 的序列。接下来p5与lane上的p5‘配对,测得了index2,并洗脱
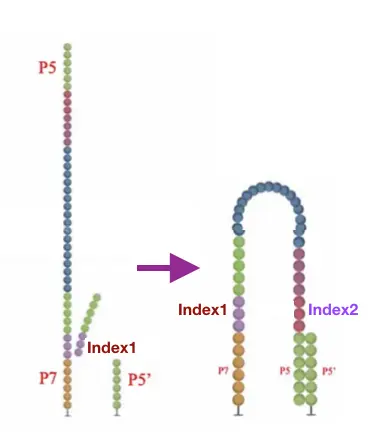
双端测序之Reverse Strand: 洗脱掉index2 产物后,还是一个桥式扩增,得到双链,再变性得到原始Forward strand 和 新的Reverse Strand, 除去测完的Forward strand。然后和测Forward一样,也是先连接primer,只是连接的位点是Primer Binding Site2,测完后得到reverse strand序列
一个小补充:知道了PE Seq,那么单端SE 测序怎么测的呢? single-end只将index,Primer binding site以及P7/P5添加到 fragamented DNA片段的一端,另一端直接连上P5/P7,将片段固定在Flowcell上桥式PCR生成DNA簇,然后单端测序读取序列
一点小问题: illunima一次只添加一个dNTP,确实能够保证准确测量同聚物的长度问题(同聚物就是3‘端一个一个加dNTP聚合而成的聚合物,因为还带接头,所以不能直接说成DNA分子)。 那么为何illumina会限制合成的链的长度呢,不能像Sanger法一样,最长测1k? 原因就出在二代测序多出来的PCR过程:比如一段时间后的PCR得到的每个cluster都各包含200条完全相同的序列,第一轮假设有199个加了碱基A测得了红色,一个没有加上就没有测得颜色;第二轮加入G应该都测得绿色,但是之前的那个应该加A,它得到了红色,这就是作为杂信号存在的。依次向下,测序长度越长,杂信号越多,最后可能标准信号和杂信号各一半,这样系统无法判断,只能给N,而N多了对于后续的分析处理很麻烦,去了吧丢失数据,不去吧又是冗余。
数据产生:
大体上我们平常使用的测序仪就是这样(以Hiseq 2000为例),后续升级版主要提高通量
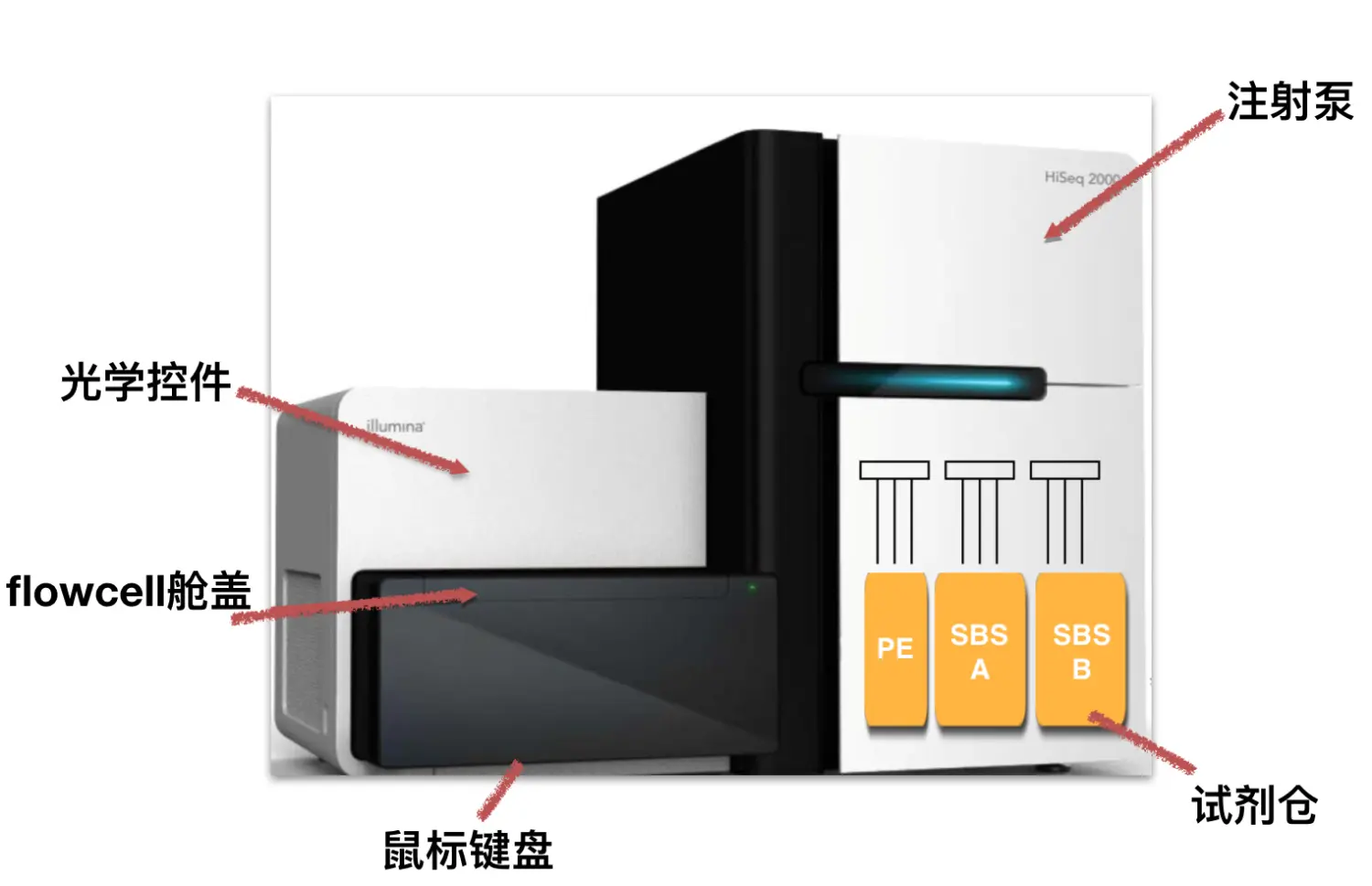
简单了解下数据产生大体流程:
从荧光信号的产生到碱基序列的识别这一过程,主要包括图象校正(即空间校正)、cluster识别、荧光校正(即光学校正)、phasing/prephasing(即化学校正)、碱基识别、PF(Illumina默认的数据过滤算法Pass Filtering)、质量评估等7个步骤
照相机是如何识别的?
利用了CCD相机(1)对每一个簇(cluster)进行识别,确定其坐标;(2)提取每个簇分别在A、G、C、T四个波长的信号强度值。另外拍照过程相当耗时,一次循环所产生的信号需要40分钟左右才能拍照收集完毕。使用相机的扫描功能会更快一些

数据量产出: 测序仪搭配了两个flowcell,简称双流动槽。比较经典的Hiseq2500一次能产出700-800Gb数据(此处Gb为测序碱基数,不同于字节数的Gb)
关于数据转换,举个例子比较好理解:Gb是测序中的数据量,1 Gigabase= 十亿碱基。人类全基因组测序得到了90G的原始数据,也就是900亿碱基。得到的900亿碱基,也对应900亿个质量值,加起来就是1800亿个字符。想一想fastq的格式:第一行是测序说明,一般是45个字符,也就是说,每一条测序reads中第一行就有大概45个字符。那么多少条reads呢?根据PE150计算:测序策略是一条reads包括150bp,现在900亿碱基,就对应900亿/150=60亿条reads 。因此第一行总字符是:60亿*45=270亿个字符。注意到fastq文件共四行,其中1、2、4行的总数量分别为270亿、900亿、900亿,第三行就是一个+,基本可以忽略不计。加起来总共2070亿字符。计算机中,根据编码规则不同,字符与字节对换关系不同。

Fastq文件是ASCII编码文件,其中每一个字符就对应一个ASCII码,也就等于一个字节。计算机的1 GB(Gigabytes) 是1024^3 个字节 因此,二者对换关系就是:全基因组测序的90Gb对应(2070x10^8 /1024^3 )=193GB计算机存储空间。 或者更快的计算: 测序报告会给出reads数,如果测序策略是PE150,那么占用硬盘空间大小就是n(reads)(150+150+45)/1024^3 另外,测序仪下机后的数据都是用gz压缩后的文件.fastq.gz,能压缩2.7倍,大概71G左右。
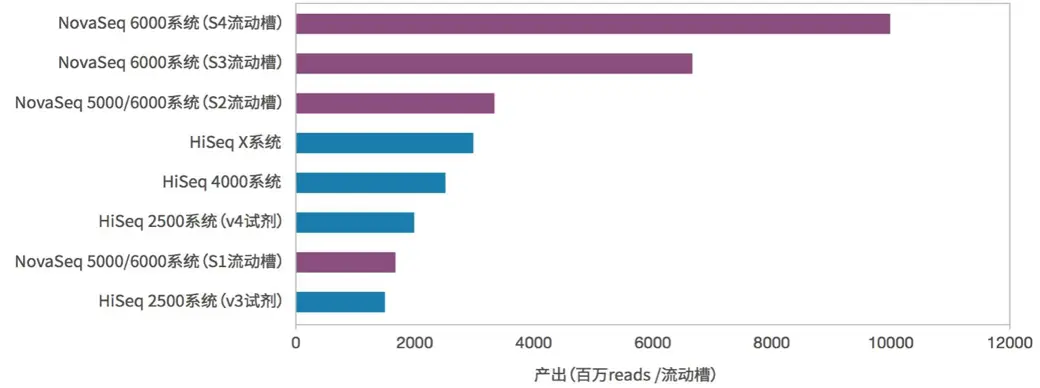
看一下来自illumina官网的统计数据,换算一下大概能知道,高配的HiSeq X10每台机器每轮测序所产生的数据量大约是1.6Tb,即1600 Gb,2017年的JP摩根大会上发布的NovaSeq 6000一次测序保守估计能得到3000G的测序数据。如果要做人类基因组重测序,Hiseq系列一般需要3-5天,而Nova只需要40小时!
数据初步分析:
使用fastqc进行质量分析,这是一款Java软件,支持多线程。写这篇文章的时候版本是v0.11.7。
软件前期准备:
- 下载方式有两种:
- 官网下载好用filezilla导入linux服务器
- 直接在服务器中wget http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.7.zip
- 接着安装
unzip fastqc_v0.11.7.zip–>cd FastQC– >chmod755 fastqc
最后这个chmod有必要说一下,这个权限管理命令
chmod 用3个数字来表达对 用户(文件或目录的所有者),用户组(同组用户),其他用户 的权限: 如:chmod 755 fastqc 数字7是表达同时具有读,写,执行权限:(7 = 4 + 2+ 1) 读取--用数字4表示; 写入--用数字2表示; 执行--用数字1表示; 三者皆否:0
设置完权限后,还需要将FastQC文件夹(这里请注意是文件夹,而非fastqc这个可执行程序)导入环境变量
echo 'export PATH=/YOUR/FASTQC PATH/:$PATH' >> ~/.bashrc再
source ~/.bashrc检查软件是否安装成功
fastqc --help出现帮助信息就可以使用啦!
后期软件使用:
基本格式 (各种参数+多个文件~支持多线程)
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam] seqfile1 .. seqfileN
-o --outdir FastQC生成的报告文件的储存路径
--extract 使用这个参数是让程序不打包【默认会打包成一个压缩文件】
-t --threads 选择程序运行的线程数,每个线程会占用250MB内存(一般与文件数量一致就好)
-q --quiet 安静运行模式【不选这个选项,程序会实时报告运行的状况】
结果分析:
- 如果你有自己的转录组或者其他数据,可以现在测试了
- 如果没有,想学一下这个软件流程以及如何解读结果,可以下载公共数据(下载两个双端测序文件共4个)
# 顺便说一下这里用到了curl -O(保留远程文件的文件名) -L(对于自动跳转的网页,curl 就会跳转到新的网址)
# 当然用wget也可以,至于二者区别,可以参考https://www.cnblogs.com/lsdb/p/7171779.html
curl -O -L https://s3-us-west-1.amazonaws.com/dib-training.ucdavis.edu/metagenomics-scripps-2016-10-12/SRR1976948_1.fastq.gz
curl -O -L https://s3-us-west-1.amazonaws.com/dib-training.ucdavis.edu/metagenomics-scripps-2016-10-12/SRR1976948_2.fastq.gz
curl -O -L https://s3-us-west-1.amazonaws.com/dib-training.ucdavis.edu/metagenomics-scripps-2016-10-12/SRR1977249_1.fastq.gz
curl -O -L https://s3-us-west-1.amazonaws.com/dib-training.ucdavis.edu/metagenomics-scripps-2016-10-12/SRR1977249_2.fastq.gz
# 下载完可以检查一下数据完整性,当然不是必须
md5sum *.gz
# 质控四个文件,我们可以采用四线程
# 大概用时 real 0m23.344s (如果你也想统计时间,就在命令前加time)
fastqc *.gz -t 4
#将结果.html用filezilla导出,浏览器查看
- 首先看到的是一个总结报告,左边这一栏会告诉你,哪些是提出警告的(⚠️表示),那些是fail的(❌)
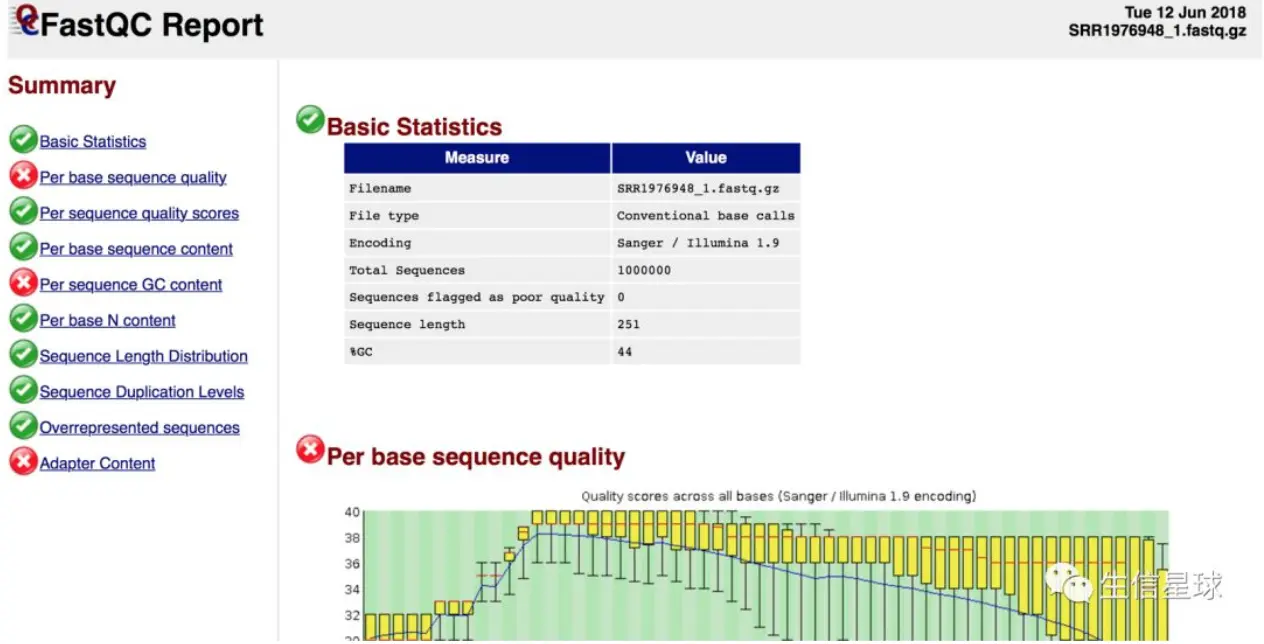
接下来一部分一部分解析,共10部分
Basic Statistics 基本信息
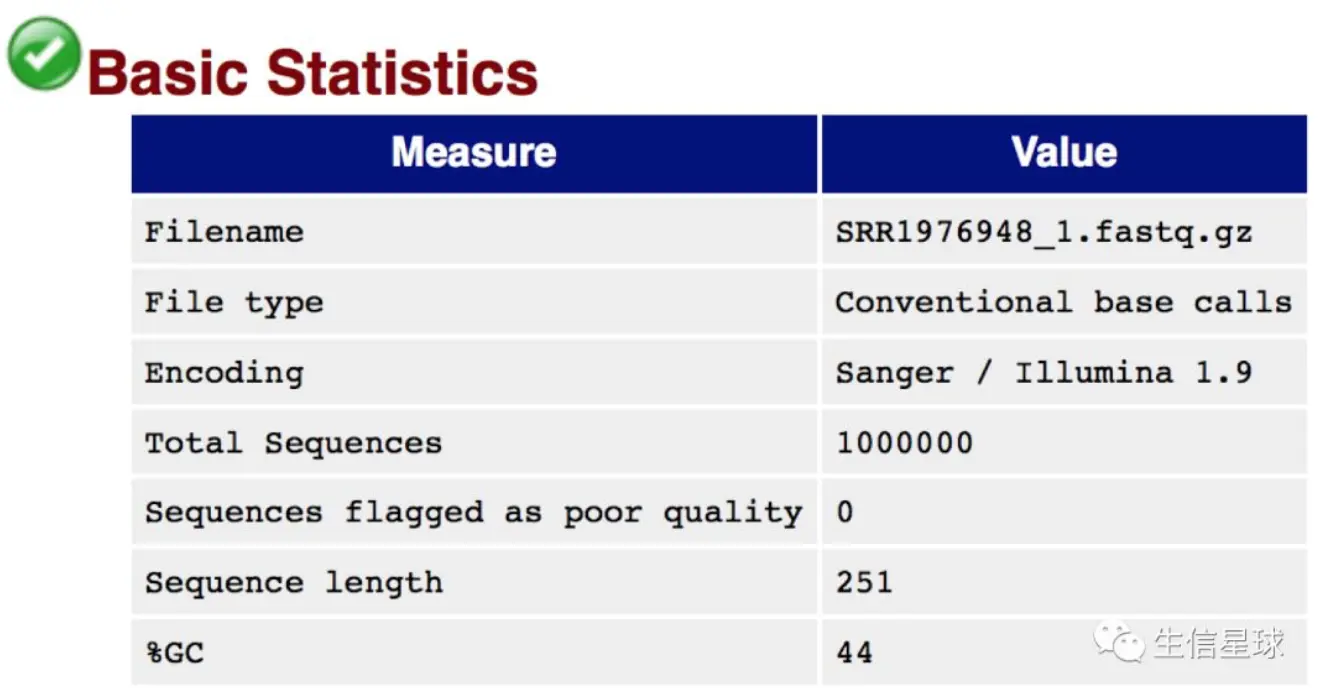
- Encoding: 测序平台编号,现在Sanger/ Illumina 1.8以上都是Phred 33编码
- Total sequences: reads数量(reads就是高通量测序平台产生的序列标签,翻译为读段!)
- Sequence length: 测序长度
- %GC: GC含量: 需要重点关注,可以帮助区别物种,人类细胞42%左右
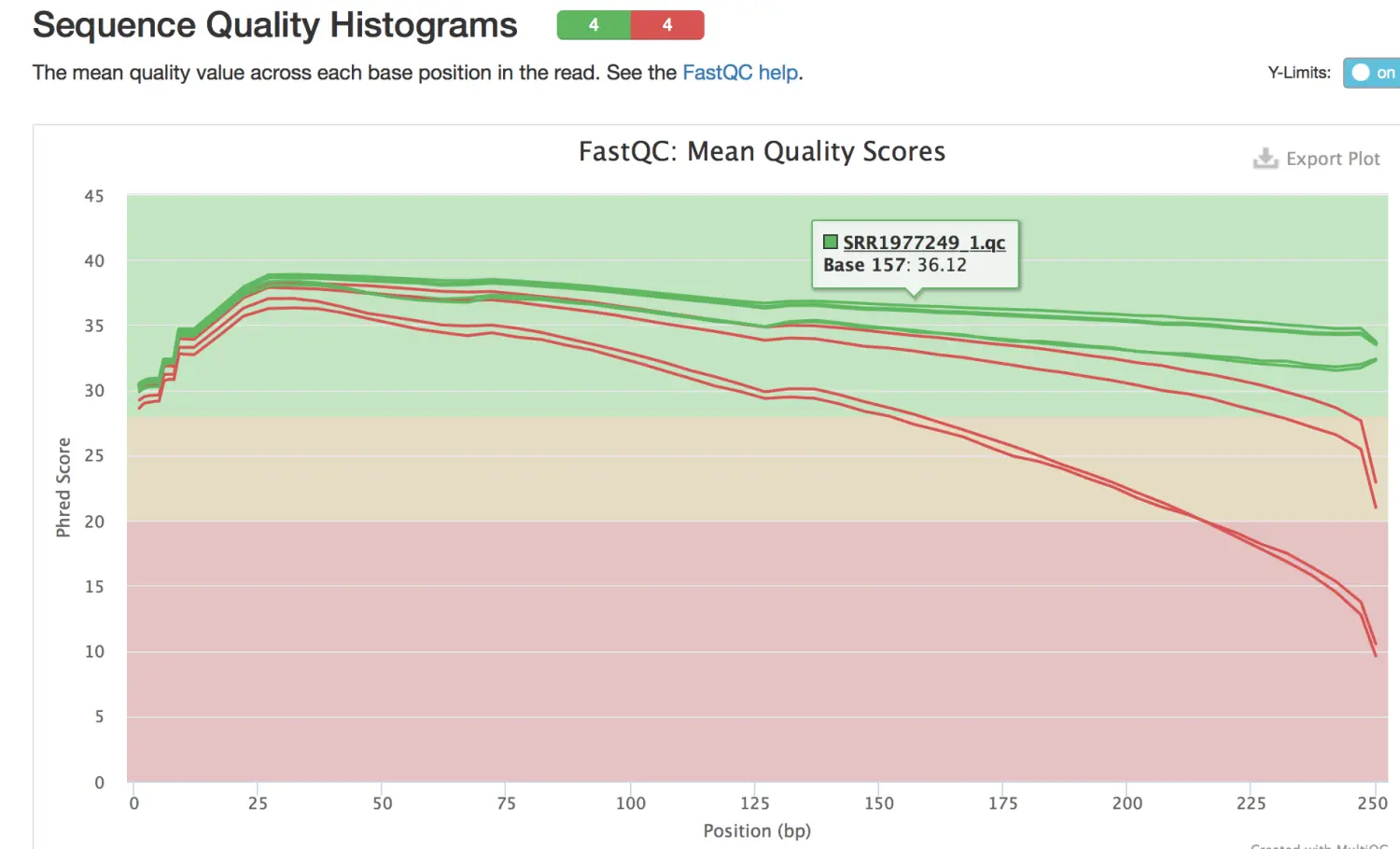
Per base sequence quility:每个测序read各碱基质量【十分重要!】
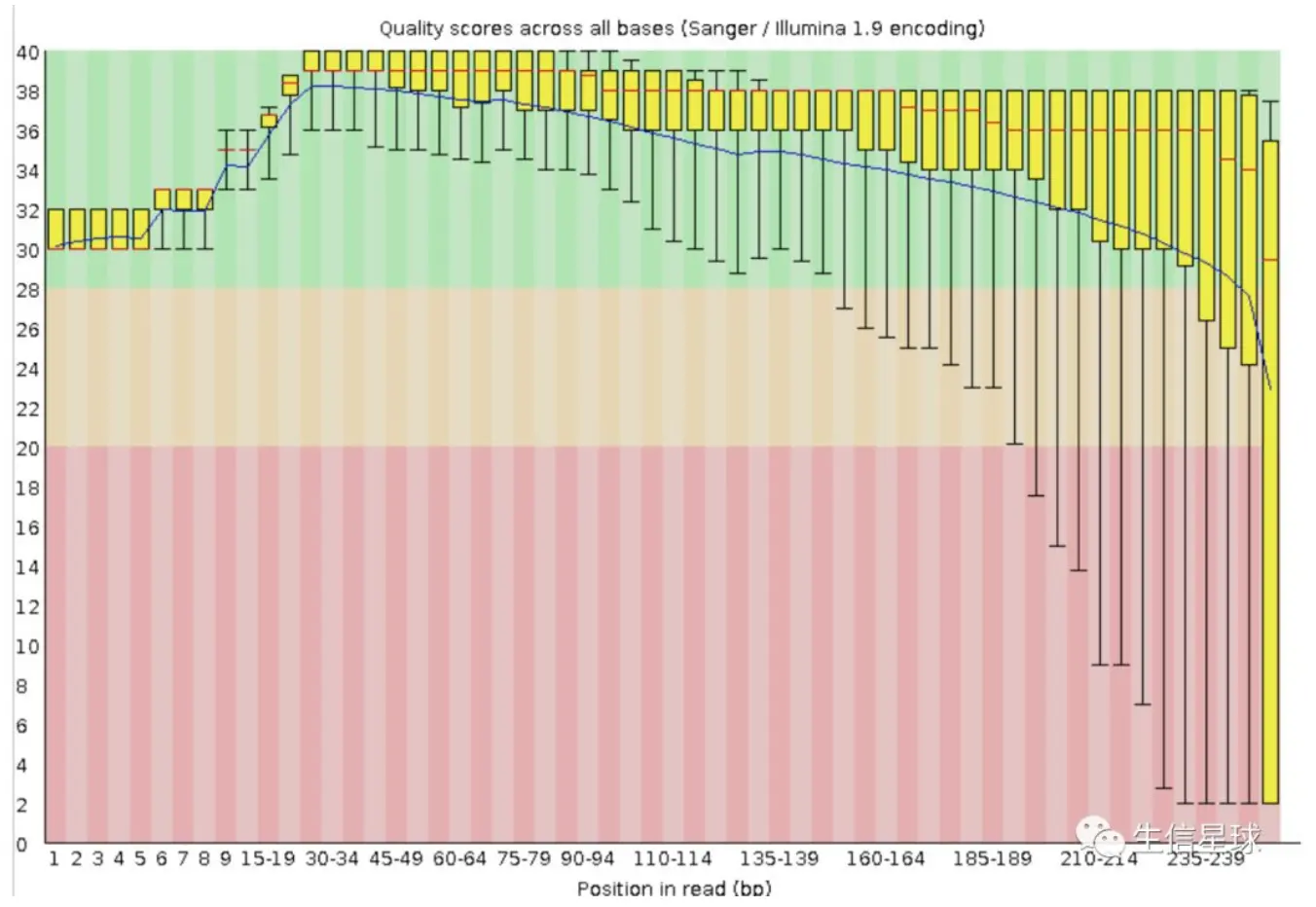
横轴:测序序列的1-251个碱基;
纵轴:质量得分,score = -10 * log10(error),例如错误率error为1%,那么算出的score就是20
箱线图boxplot:对每一个碱基的质量的统计。箱子上面的须(up bar)为90%分位数,下面的须(down bar)为10%分位数,箱子中的红线为中位数即50%分位数,箱子顶(upside)为75%分位数,箱子低(downside)为25%分位数。这个boxplot的意义:一是看数据是否具有对称性;二是看数据分布差异,这里主要利用了第二点。bar的跨度越大,说明数据越不稳定。
蓝色的线将各个碱基的质量平均值连接起来
解释一下:图中蓝线的走势为何先高后低? 因为目前采用的边合成边测序使用的是化学方法促使链由5’向3’延伸,也就是利用了DNA聚合酶。 刚开始测序,合成反应还不是很稳定,但是酶的质量还很好,所以会在高质量区域内有一定的波动(这里的1-30bp),后来稳定了,但是随着时间的推移,酶的活力逐渐下降,特异性也变差,所以越往后出错几率越大。【就像一个司机开车,一开始小心谨慎,起步慢,开的也慢,慢慢提速。后来越开越带劲,但是也越来越困,疲劳驾驶容易出事】
一般能用的数据都要求至少Q20,也就是下四分位(10%分位数)的质量值要大于20。因此这里的189bp后面的需要切除,才能继续分析
二代测序,最好是达到Q20的碱基要在95%以上(最差不低于90%),Q30要求大于85%(最差也不要低于80%)
Per sequence quility scores:每条序列 质量统计
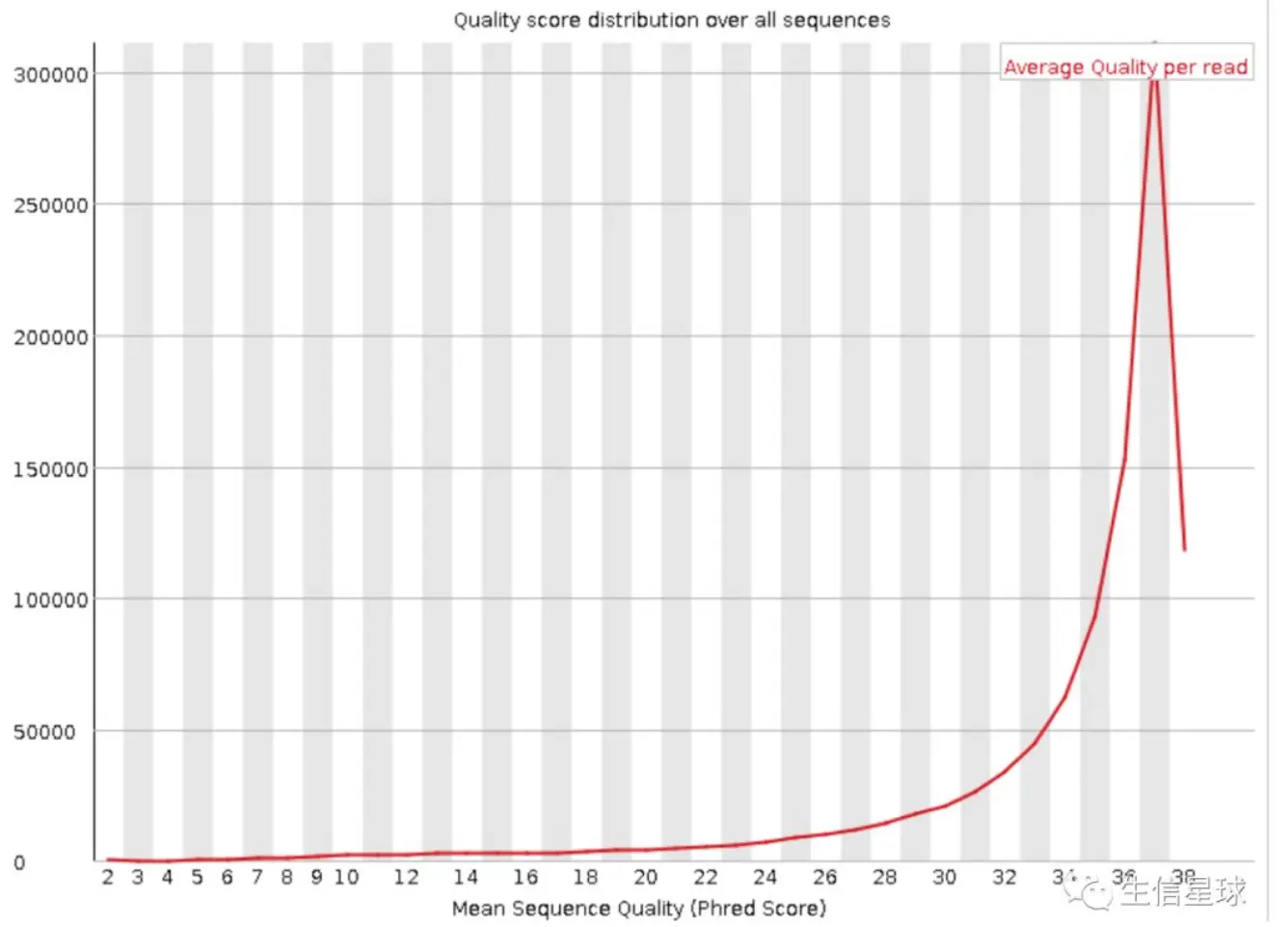
横轴:质量值0-40,也即是Q值
纵轴:每个质量值对应的read数
我们的例子中一条read有251bp, 那么其中任意一条的251bp的质量平均值就是这条read的质量值。只要大部分都高于20说明比较正常
Per base sequence content:read各个位置碱基比例分布
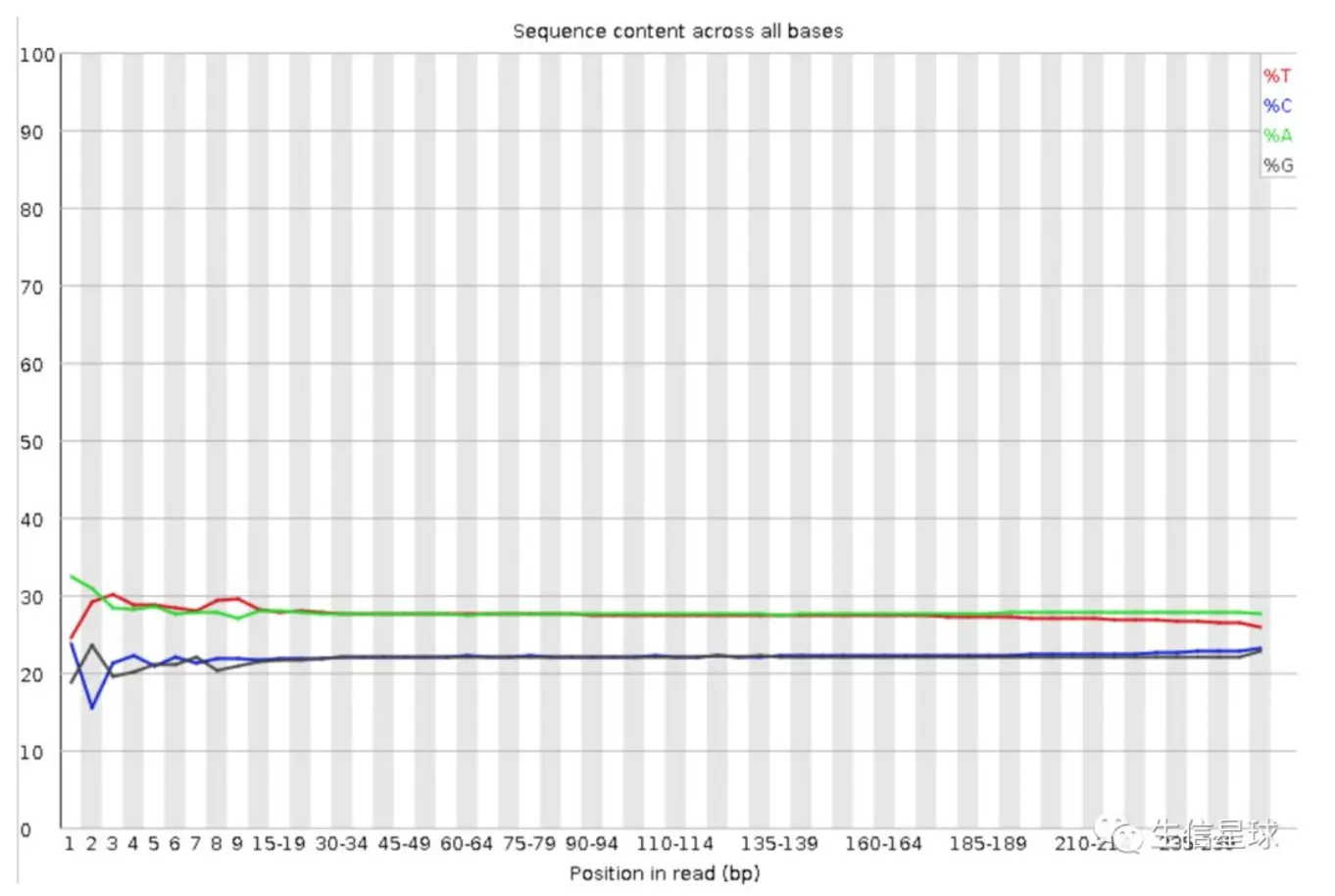
横轴:各碱基位置;纵轴:碱基百分比
四条线四种颜色代表四种碱基在每个位置的平均含量(一个位置会测很多reads,然后求一个平均)
一般来讲,A=T, C=G, 但是刚开始测序仪不稳定可能出现波动,这是正常的。一般不是波动特别大的,像这里cut掉前5bp就够了。另外如果A、T 或 C、G间出现偏差,只要在1%以内都是可以接受的
Per sequence GC content: 序列平均GC分布
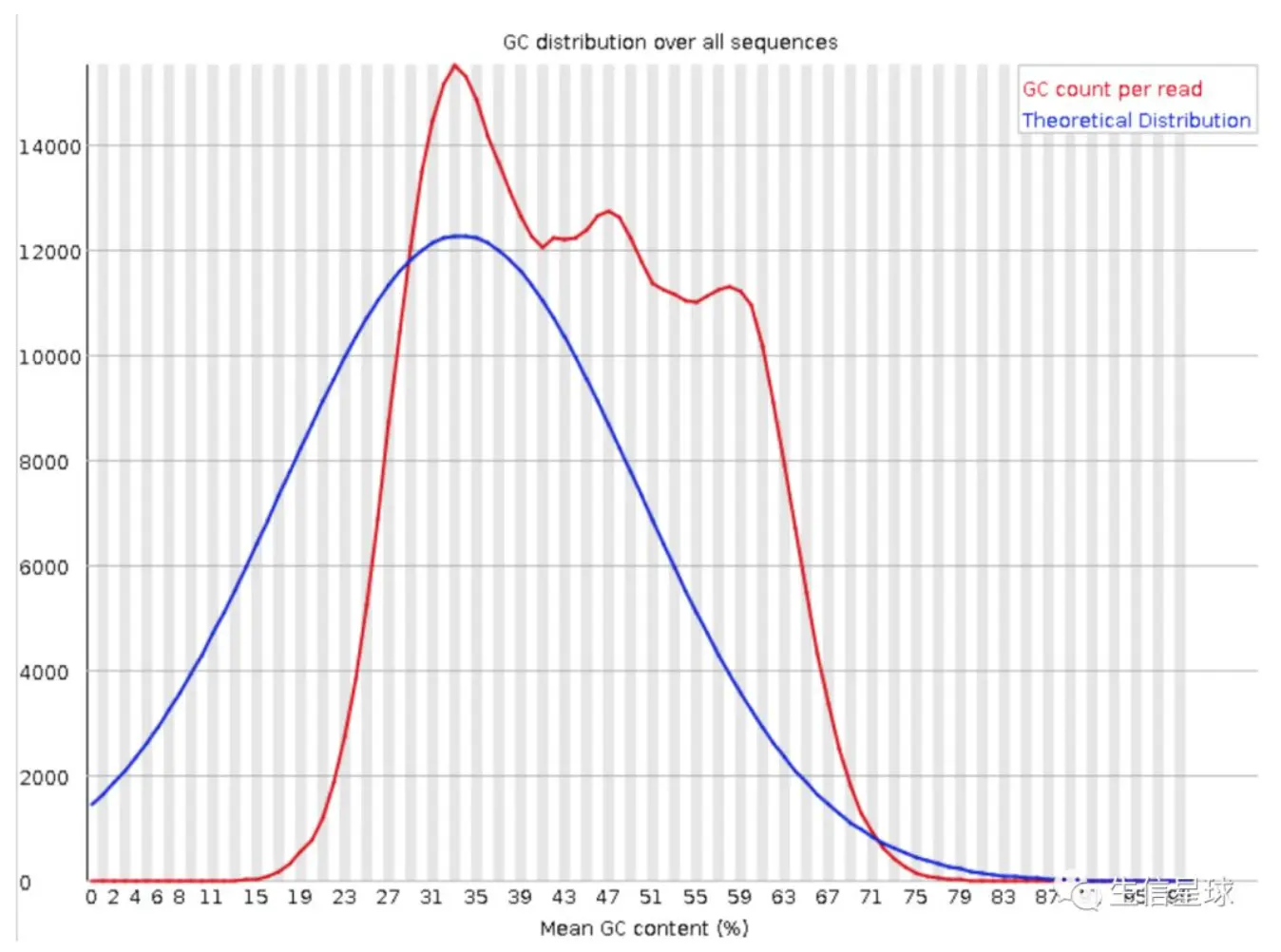
- 横轴为平均GC含量; 纵轴为每个GC含量对应的序列数量
蓝线为系统计算得到的理论分布;红线为测量值,二者越接近越好
- 这里不相符可能有两个原因:
- 前面提到了,GC可以作为物种特异性根据,这里出现了其他的峰有可能混入了其他物种的DNA; 2. 目前二代测序基本都会有序列偏向性(所说的 bias),也就是某些特定区域会被反复测序,以至于高于正常水平,变相说明测序过程不够随机。这种现象会对以后的变异检测以及CNV分析造成影响
- 如果出现怎么办?– 把和我们使用物种GC-content有差异的reads拿出来做blast,来确认是否为某些杂菌
Per base N content: N含量分布
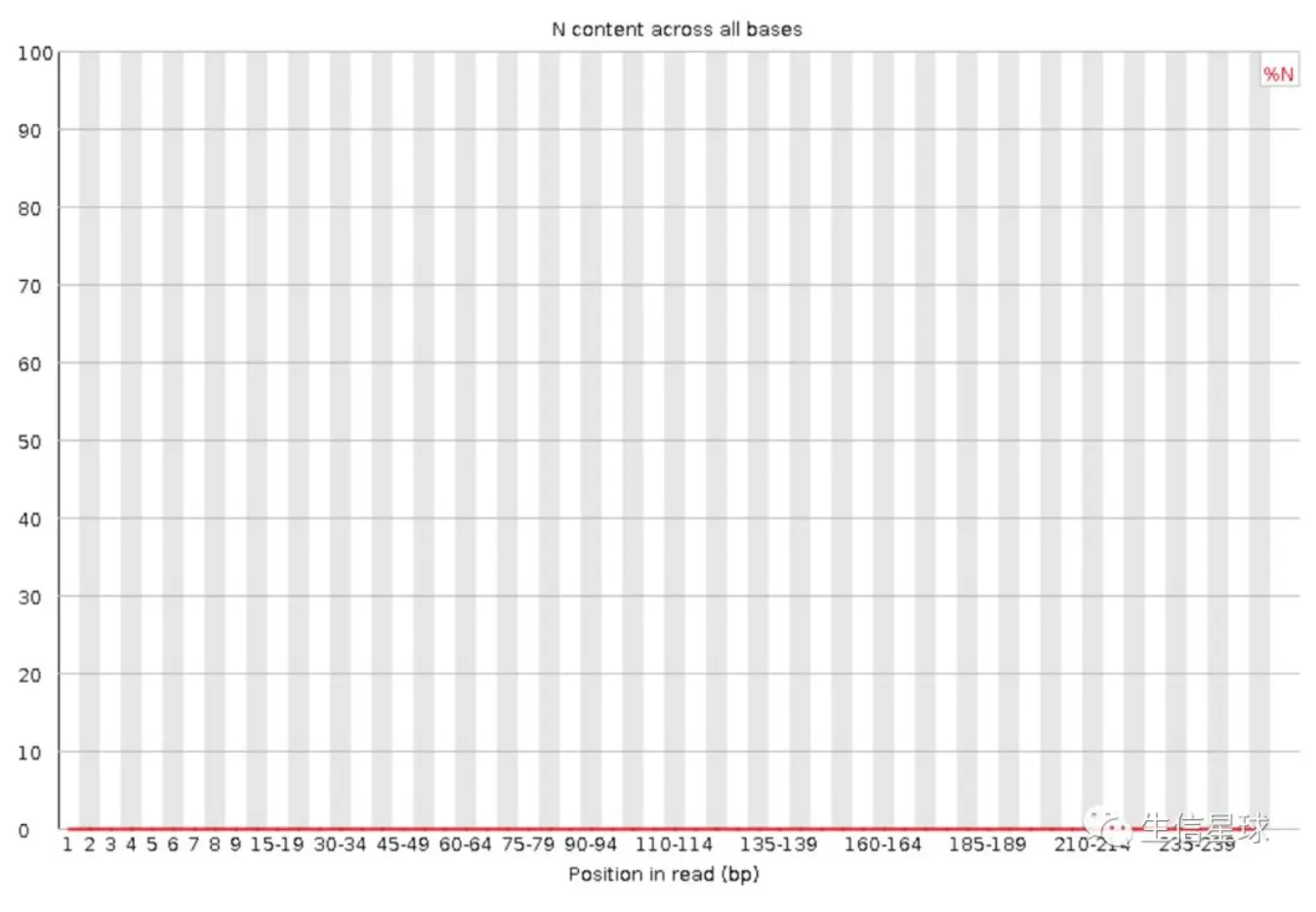
N是指仪器不能识别ATCG时给出的结果,一般不会出现。但是如果出现并且量还很大,应该就是测序系统或者试剂的问题
任意位置的N的比例超过5%,报"WARN”;任意位置的N的比例超过20%,报"FAIL”
Sequence length distribution: 序列长度统计
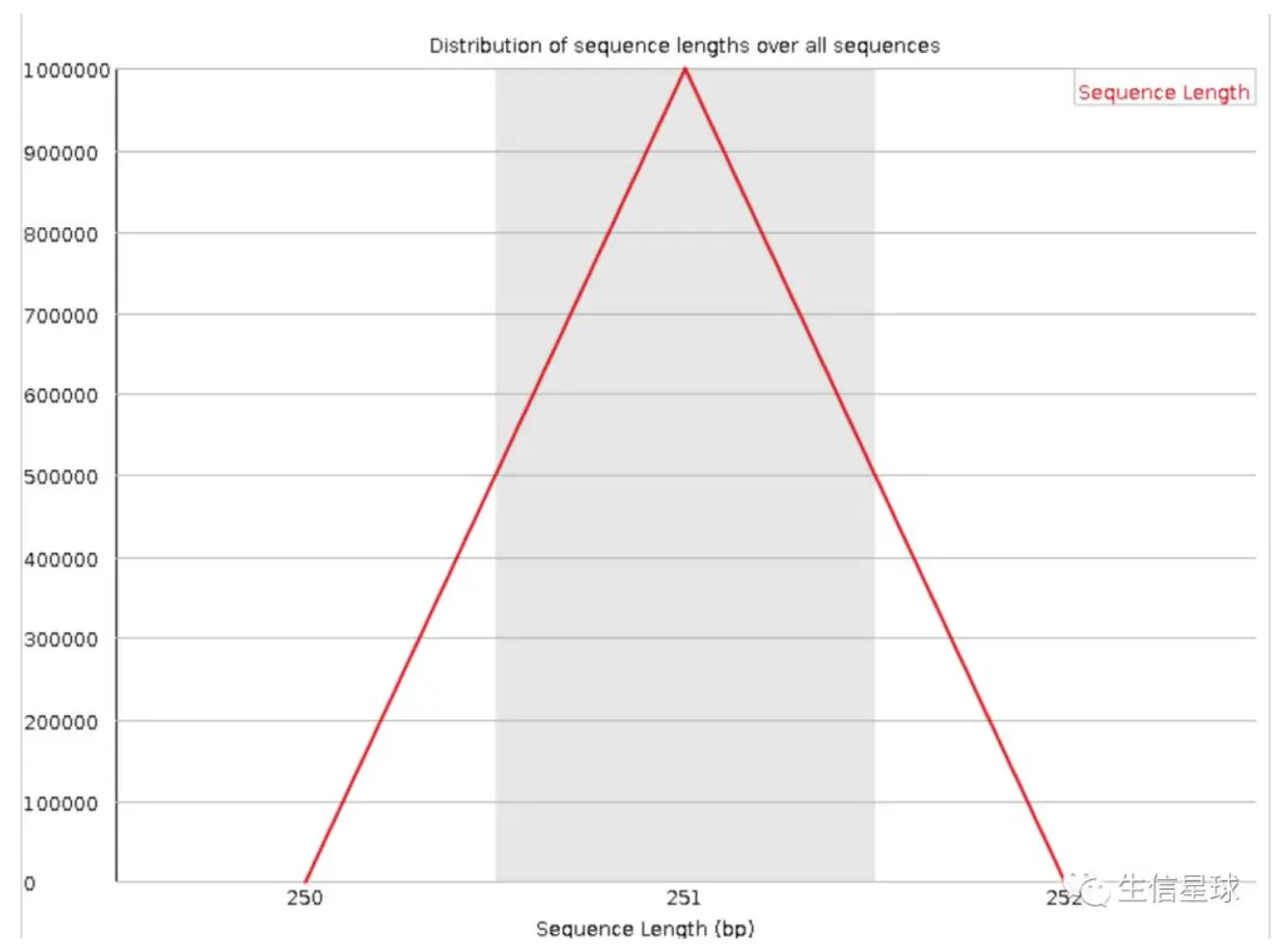
理想情况下,测得的序列长度应该是相等的。实际上总有些偏差
当reads长度不一致时报"WARN”;当有长度为0的read时报“FAIL”
这里显示大部分都落在251bp这个测序长度上,有少量为250或252bp,但这不影响;如果偏差很大就不可信了
Sequence duplication level:统计序列完全一样的reads的频率
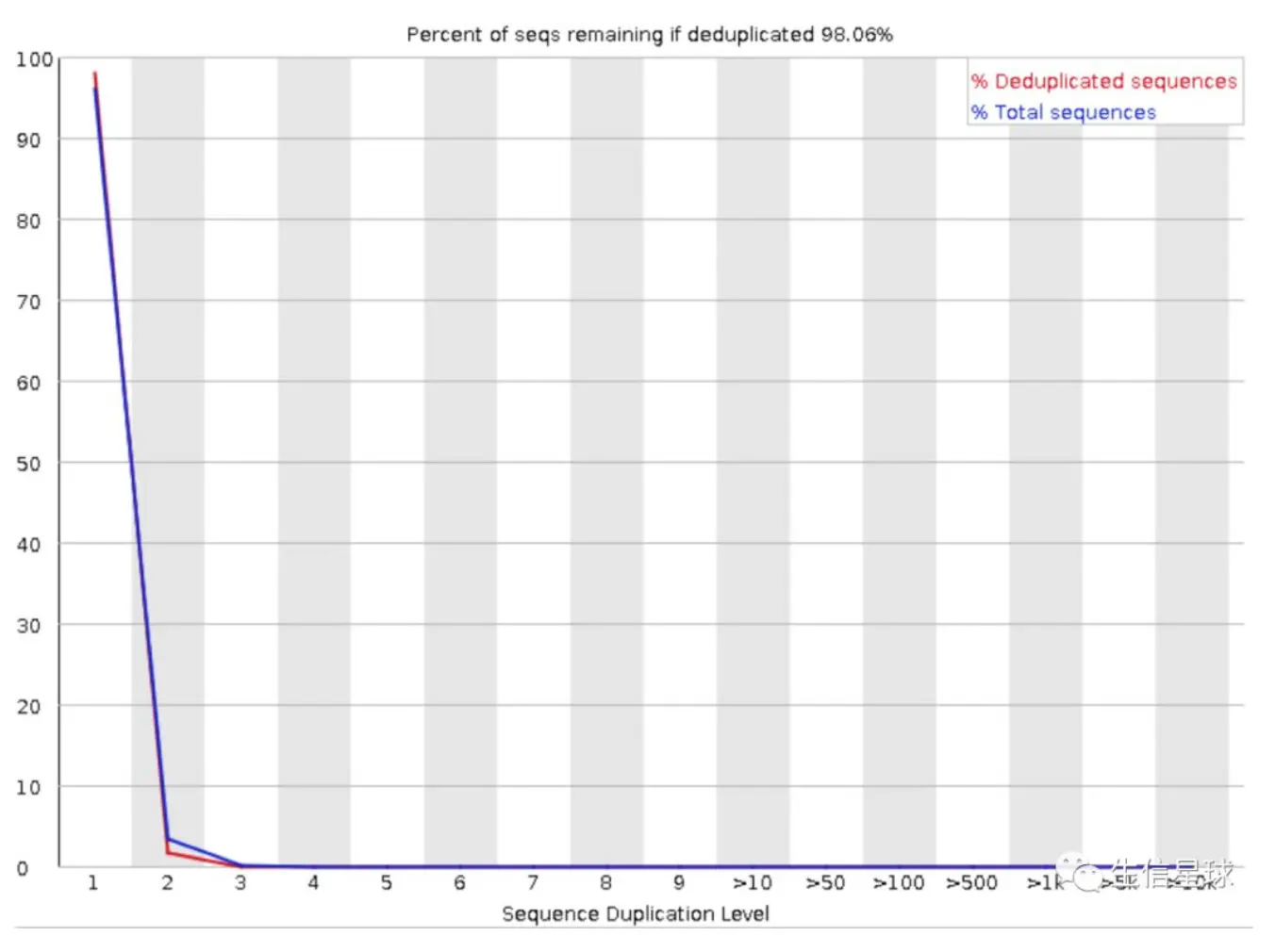
横坐标是duplication的次数;纵坐标是duplicated reads的数目(红线)
解释下横坐标为何会有>10, >50等出现: 测序的原始数据很大,如果每一条reads都统计,将耗时很久。 这里软件只采用了数据的前200,000条reads统计其在全部数据中的重复数目,另外大于75bp的reads只取50bp进行比较。重复数大于10的reads被合并统计成了>10,以此类推…
unique reads总数(蓝线)作为100%,上图中可以看出,大概仅有2%的uniqe reads可以观察到两次重复。也就是说,我们这里的非unique reads占总数比例仅有2%左右。
正常情况下的确,测序深度越高,越容易产生一定程度的duplication。高程度的duplication level,提示我们可能有bias的存在(如建库过程中的PCR duplication)。
另外和做的项目也有关,一般转录组测序的结果中duplication level都比较高,60-70%都正常,这是因为转录组测的是基因的覆盖深度,各个基因表达量不同,如果某个基因覆盖度较高【tip:覆盖度是指基因/转录组测序测到的部分占整个组的比例】,那么测的部分就越多,相对应的duplication也会更高;对于外显子组测序来说,一般覆盖度比较一致,这里出现了duplication就不太正常。
当非unique的reads占总数的比例大于20%时,报"WARN”;当非unique的reads占总数的比例大于50%时,报"FAIL“
Overrepresented sequences:大量重复序列

和第8个duplication计算一样,也是取前200,000进行统计,大于75bp只取50bp。
发现超过总reads数0.1%的reads时报”WARN“,当发现超过总reads数1%的reads时报”FAIL“
Adapter content: 接头含量

表示序列中两端adapter的情况
软件内置了四种常用的测序接头序列, fastqc 有一个参数-a可以自定义接头序列
此图中使用的illumina universal adapter并未去除,后期再使用trimmomatic/cutadapt去接头
我们在学习测序原理的时候知道了,接头的作用一个是能够使得序列结合到flowcell上,另外一个多样本测序时利用接头旁边的index加以区分
什么情况下能够测到接头呢? 一般测序read的长度大于插入片段(待测序列)的长度时会发生。对于WGS建库测序来讲,一般不会发生,因为他的待测序列要几百bp,而测序也就150bp算高了。但是对于RNA-seq,一般测序序列比较短,尤其是miRNA,只有几十bp,这是就会测到read末尾的接头
(还有一类这里没体现)Kmer content: 重复短序列
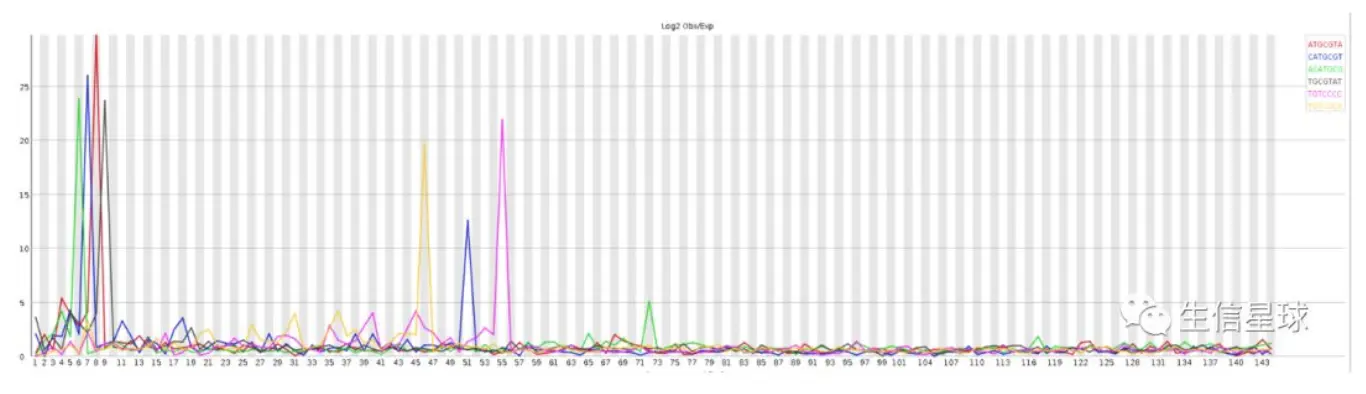
- 表示:在序列中某些特征的短序列重复出现的次数
* 这个图是转录组测序的一个文件,可以看到6-9bp几种短序列都出现了好多次。**出现的原因**可能是:
1. 没有去除软件内置的adapter或者没有使用-a参数自定义adapter
2. 序列本身重复度较高,例如在建库PCR过程出现序列偏向性bias--> 这在转录组测序中确实存在
数据过滤:
主要针对接头序列和低质量序列
工具: 有许多工具能干这事,例如SOAPnuke、cutadapt、untrimmed、sickle和seqtk等,其中经常用到的是Trimmomatic(也是一个java程序)
Trimmomatic:
安装: 官网下载我使用的是0.36版本

下载后直接解压安装,其中有一个trimmomatic-0.36.jar是执行文件,使用时输入java -jar trimmomatic-0.36.jar就可以。另外还有一个adapter文件夹,里面存放了常用的illumina测序仪接头序列fasta格式,后续处理接头需要制定具体文件。
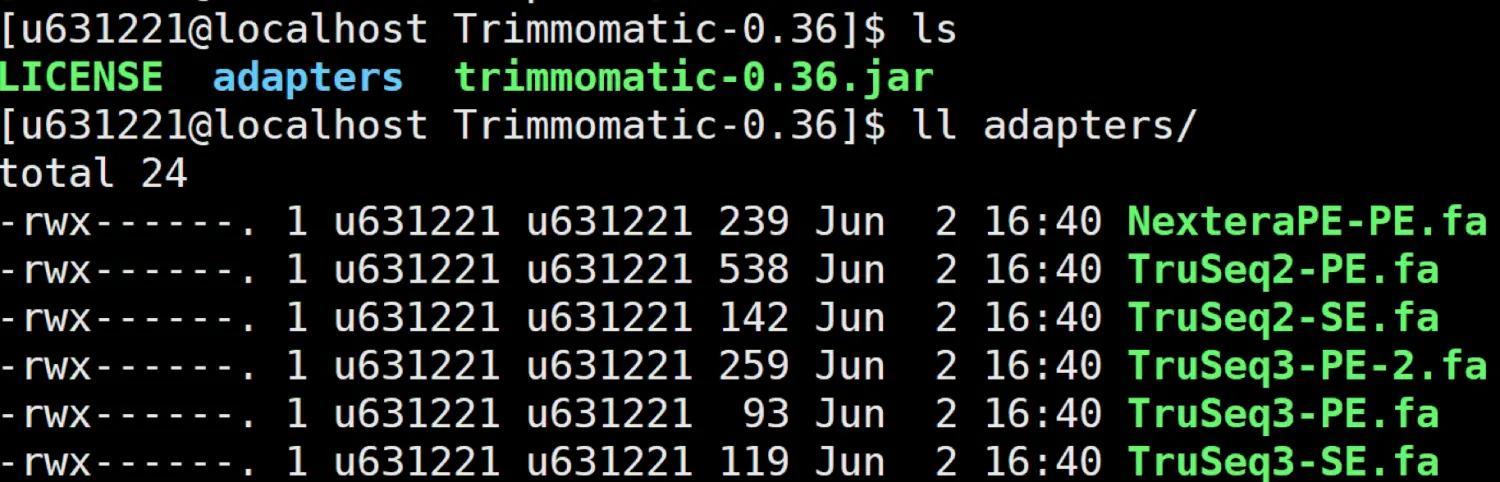
关于adapter: 目前绝大部分的illumina的Hiseq和Miseq系列使用的都是Truseq3,过去的GA2测序仪使用的是Truseq2,PE/SE对应单端还是双端测序 , 如果使用的不是illumina测的,可以按照里面的格式自己新建一个接头文件,但其中的命名要注意。详情见官网主页

以PE测序为例说一下命令参数设置:
java -jar <path to trimmomatic.jar> PE [-threads <threads 线程数] [-phred33 | -phred64] 质量体系,默认64,但我们现在一般都是33 [-trimlog <logFile>] log文件 <input 1> <input 2> 双端测序原始fq文件 <paired output 1> <unpaired output 1> 双端1输出文件 、 过滤掉的文件 <paired output 2> <unpaired output 2> 双端2同理 <step > <详细讲一下step:> 1. ILLUMINACLIP: 根据上面qc部分的测试数据,我们设置TruSeq2-PE.fa:2:40:15 TruSeq2-PE.fa是接头序列; 2是比对时接头序列时所允许的最大错误数; 40是要求PE的两条read同时和adapter序列比对,匹配度加起来超40%,那么就认为这对PE reads含 有adapter,并在对应的位置需要进行切除;【那么为何不是匹配100%?因为测序时并不是把 adapter全测了,只测了一部分】 15 指的是只要某条read的某部分与adapter超过了15%的相似度就认为包含,就要去除 2. SLIDINGWINDOW: 滑动窗口长度 我们设置为4:20,代表窗口长度为4,窗口中的平均质量值至少为20,否 则会开始切除 3. LEADING,指的是read开头的碱基是否要被切除的质量阈值,这里设为2 4. TRAILING,指的是read末尾的碱基是否要被切除的质量阈值,这里设为2 5. MINLEN,指的是read被切除后至少需要保留的长度,如果低于该长度,会被丢掉,这里设置25#一个☝️范例(这个测试数据是phred 64的,所以使用默认值就好): java -jar trimmomatic-0.36.jar PE -threads 8 \ -trimlog logfile \ reads_1.fq.gz reads_2.fq.gz \ out.read_1.fq.gz out.trim.read_1.fq.gz \ out.read_2.fq.gz out.trim.read_2.fq.gz \ ILLUMINACLIP:/path/Trimmomatic/adapters/TruSeq2-PE.fa:2:40:15 \ SLIDINGWINDOW:4:20 \ LEADING:2 TRAILING:2 \ MINLEN:25# 当然,这只是对一个样本的双端测序文件进行操作,那么问题来了: # 如果你的样本比较多呢?还要手动一个个输入吗? # 虽然说vim中使用快速替换 :%s/AA/BB/g 可以全局替换AA成BB,但还是有点麻烦 # 能不能让程序来一个自动化呢?是可以的!可以看作是上面👆脚本的改进版~ # 在脚本中构建for循环 # vi trim.sh #开头这样写而不写*.gz的目的是避免了样本名称的重复 for filename in *_1.fastq.gz do #对于filename(%)向右匹配_的全部内容(*),然后这部分去掉,留下这之前的 base=${filename%_*} java -jar trimmomatic-0.36.jar PE \ # 加上反斜线能让程序整体更清晰 -threads 8 \ -trimlog logfile \ ${base}_1.fastq.gz ${base}_2.fastq.gz \ out.${base}_1.fastq.gz out.trim.${base}_1.fastq.gz \ out.${base}_2.fastq.gz out.trim.${base}_2.fastq.gz \ ILLUMINACLIP:/home/u1239/biosoft/Trimmomatic-0.36/adapters/TruSeq2-PE.fa:2:30:10 \ SLIDINGWINDOW:5:20 \ LEADING:5 TRAILING:5 \ MINLEN:50 done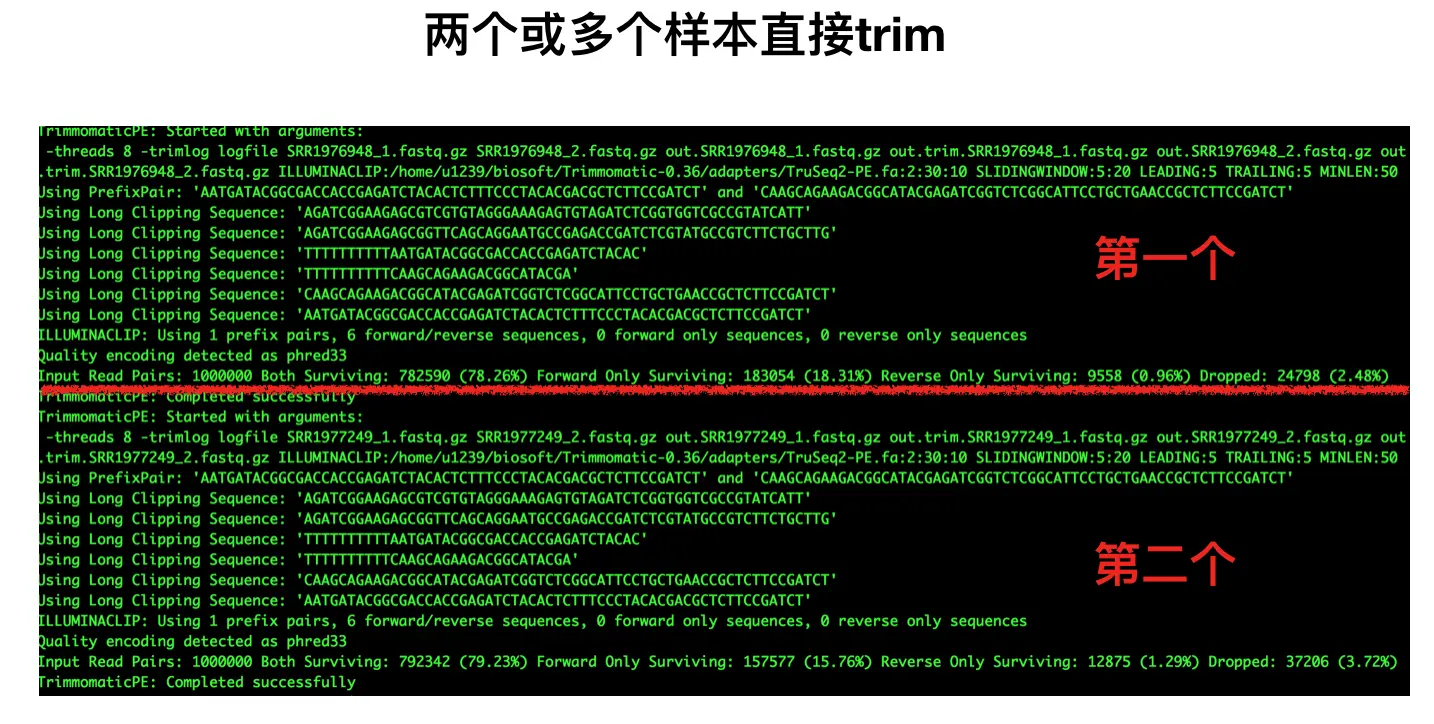
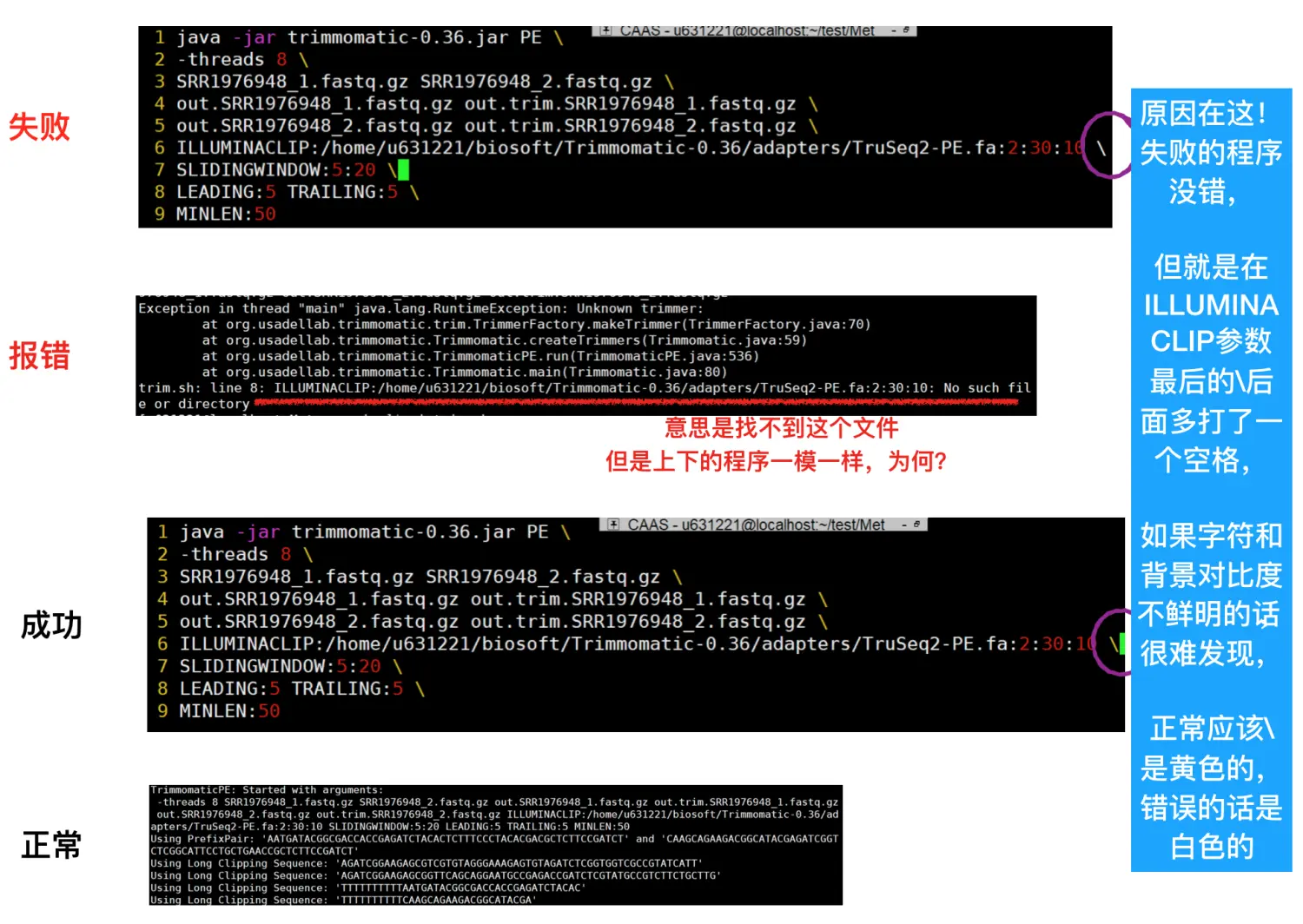
如果报错,就说明参数错误 例如我在运行的时候就有的问题百思不得解,敲了好几遍代码应该没错,但就是报错,换个服务器就好了,难道是平台问题?其实并不是,就是因为一点点小地方,有时只是一个代表换行的反斜线
\
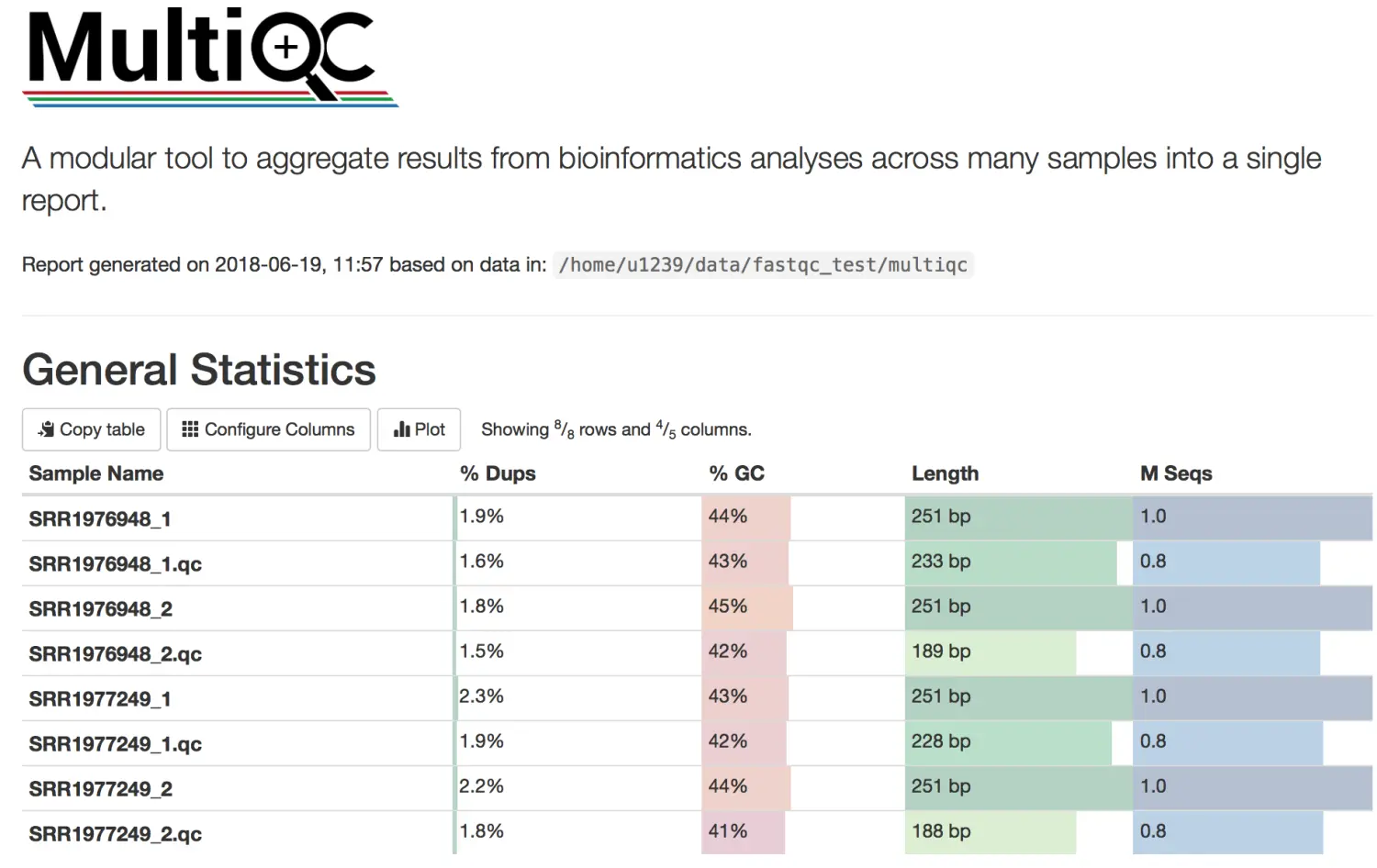
多样本质控:
fastqc对于一两个样品还能吃得消,但样本多了,我们如何同时查看对比他们的信息呢?
这里就可以使用界面更加优美的multiqc软件了, 他就是fastqc结果的整合 我会从一个初学者角度来一步步进行,其中包括了中间犯的错误及改正~,如果你也在运行的过程发现了类似的错误,可以参考一下。
分界线1: 尝试自己安装
下载地址:
解压缩后,会发现和以前安装的源代码文件不同,他没有直接显示可执行文件,也没有配置文件。这是因为multiqc是一个python软件,这里先看一下setup.py:
直接使用python setup.py会报错,因为可能你的服务器并没有setuptools,这是python依赖的第三方库。
先安装setuptools:
下载地址:
解压缩完setuptools会看到有这些文件,这也是一般的python软件常见的:包括了setup.py,easy_install.py等
# 主要使用setup.py
python setup.py build # 编译
python setup.py install #安装
在第二步安装的时候报错,原因是不能对/usr/local/lib下的python进行操作,因为不管/usr/bin还是/usr/local/bin,都是可读不可改,如果自己家目录环境变量中查不到python的路径,那么安装过程中会自动调取更上层目录的python使用。这往往会引发一系列问题。如果要自己更改的话,需要自己home目录下有python
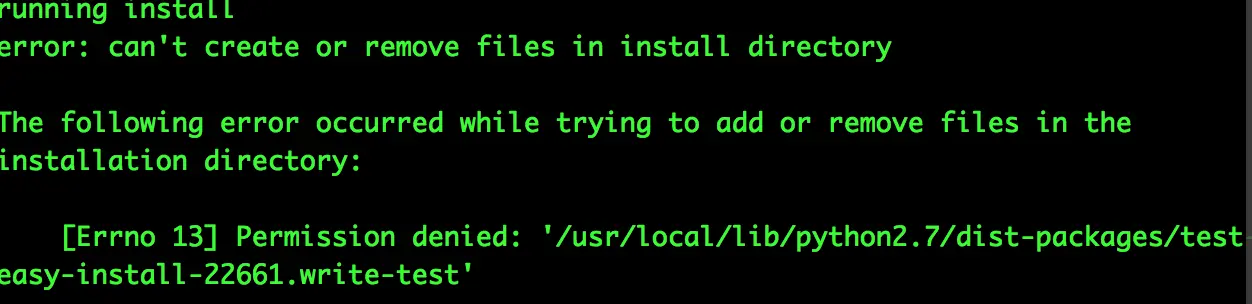
好吧,那么接下来我们再安装自己的python,毕竟自己的软件用起来方便:
# 下载地址
wget https://www.python.org/ftp/python/3.6.5/Python-3.6.5.tgz
# .tgz 就等同tar.gz
# 解压后会看到配置文件configure,设置成自己的软件环境变量即可
./configure --prefix=/YOUR PATH/
make
make install
# 可能结尾会有报错,但是不影响使用,出现了可执行文件python就成功了
# 将python复制到你的环境变量中就能直接调取
# 最后使用which python检测是否安在了自己的目录下
自己的python安装完成了,进入到解压好的multiqc文件夹下, python setup.py build 编译会运行成功,然后python setup.py install
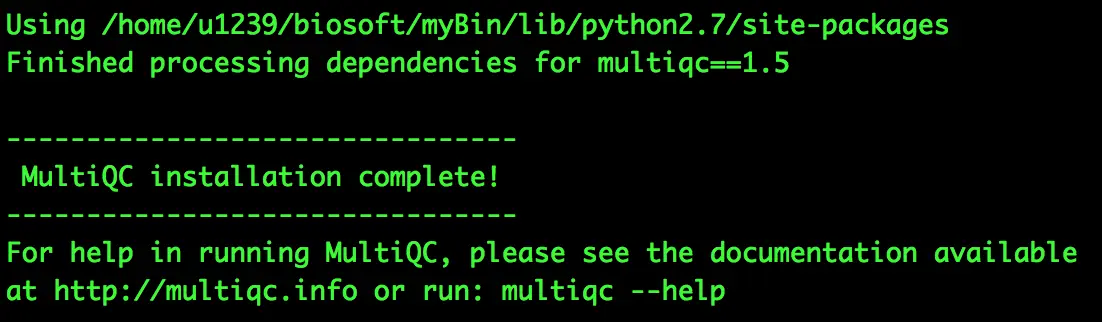
接着,multiqc就会出现在了自己的软件环境变量中了
分界线2: 同一件事,换个玩法
目的:安装multiqc
途径:conda自动安装
缘由:好久不用conda安装软件,一直坚持源代码,因为好掌控。但是有的软件依赖的包很多,自己又很难发现这些潜在的关系,所以想重新试试conda,但并不是傻瓜式的使用,而是让conda听我的话~授之以鱼,不如授之以渔
Here we go!
安装: 安装过程需要注意
- 可以自定义新文件夹,默认是在home/miniconda3/
- ⚠️不要将conda添加到PATH中,我们只需要把它视作一个保姆babysitter,而不要当一个管家housekeeper
- 安装完conda,把miniconda/bin下的conda复制到你的/biotools/PATH(这个环境变量因人而异)
添加源: 清华源和中科大源都不错,别忘了再添加一个bioconda源
新建conda专属下载目录: 你可以在你的biotools目录下新建一个conda软件存放目录,例如conda_install。然后把这个文件夹添加到环境变量。以后你用conda安装的所有软件都存放在这里

⚠️和你自己安装的软件要区分开, 然后利用conda install -p /PATH/conda_install multiqc 就可以实现multiqc的安装了
写在后面
- multiqc的使用很简单,然后结果还是交互式的,能导出很多格式
- 这次实现了人工和自动二者的有机结合:复杂的,多依赖性的软件还是要靠conda解决。但是如果一个软件用两种方法都安装了,怎么选择? 这个利用bashrc中设置的顺序:哪个目录优先,就先搜索哪个目录
结果一目了然:trim掉低质量序列和接头后,新的数据质量值都在q30以上