093-Pathway通路与GSEA基因集有何区别?
刘小泽写于19.3.19
通常做富集分析时,我们会遇到两种方法Pathway和GSEA,它们到底有什么区别?
前言
我们通过差异分析会得到一些差异基因,那么如何知道这些基因是做什么的,都影响了处理组哪些过程?Pathway和GSEA都是为了说明这个问题的。但是它们之间存在着很大的不同。因为之前我也是存在疑惑,今天听人讲起,于是查了一下
原文在此:https://advaitabio.com/ipathwayguide/pathway-analysis-vs-gene-set-analysis/ 另外结合了一下自己的认识
关于pathway
Wiki的解释:A series of interactions among molecules in a cell that leads to a certain product or a change in a cell
pathway主要描述了一种机理或者现象,可以有信号通路、代谢通路等等,它的结果由点(nodes)和线(edges)组成,目的是描述某些现象、相互作用和依赖性。Pathway是一种描述细胞、组织或个体内的基因、蛋白或代谢产物互作关系的模型,并不是简单地基因列表。我们都知道有KEGG是做富集分析通路注释的,但是还有一些数据库,比如Reactome、Biocarta等也可以做pathway分析
关于GSEA
GSEA方法由Broad Institute提出的富集方法,核心是基因集(gene set),它就是无序、无结构的一组基因,我们可以将这些基因定义成参与特定生物过程(例如:细胞周期)、存在于某个位置(例如:1号染色体)、与什么疾病有关(例如:乳腺癌),或者直接可以取某个pathway中存在的一些就因(例如:参与KEGG细胞周期通路的128个基因)。可以看到,基因集除了仅仅包含了一些基因以外,似乎没有什么定义,也正因为这样,基因集的定义可以更加广泛,主要看人为需求。
Molecular Signatures Database ( MSigDB)数据库中就包含了超过17,000个这样的基因集分布在8大类别中(如:H: hallmark gene sets、C1: positional gene sets、C2:curated gene sets、C3 : motif gene sets、C4 : computational gene sets、C5 : GO gene sets、C6 : oncogenic signatures、C7 : immunologic signatures),让富集分析不仅可以从GO、KEGG这样的功能角度出发,还可以结合位置、表达量变化趋势等进行研究,更加拓展了富集分析的范畴。
有6种情况需要pathway分析更多
第一种:想知道基因是怎么相互作用时
上面👆说到了,pathway与基因集之间一个关键的不同就是基因集是无序的,而pathway是用于描述某个过程、机制或者现象的复杂模型。

左边👈的图【KEGG MAPK pathway 】画出了各种基因和基因产物(胞内/外/膜内)的位置、互作类型(激活、抑制、磷酸化等)、信号传递方向等等;右图【MSigDB gene set corresponding to the KEGG MAPK pathway 】只是可以让我们知道有这些基因。
第二种:想充分利用差异基因在不同样本表达量改变的大小和方向时
早期的基因集分析方法是采用**ORA(Over-Representation Analysis)**的方法,一系列差异基因作为输入,然后看这个列表中的基因是过表达还是低表达(比如可以看logFC值,FC=处理组表达量/对照组表达量)。这个需要事先定义一个阈值用来决定哪个基因作为差异基因(就像定义logFC=2还是等于1.5,都是人为定义的)。然后基于每个pathway中DE (Differentially expressed)基因的富集程度来评估每种pathway的重要性,富集程度越高的pathway更可能与给定的条件真正相关。简而言之,这种方法很大程度上依赖于定义DE的标准,包括统计方法和阈值的选择。
**二代方法FCS(Functional Class Scoring )**是基于所有基因表达量来消除DE选择标准的依赖性,其背后的假设是:除了表达量变化比较大的基因以外,还有一些变化小却可能其协同作用的基因也是重要的。主要方法包括:GSEA [25], Catmap [3], GlobalTest [10], sigPathway [28], SAFE [2], GSA [7], Category [17], PADOG [26], PCOT2 [19], FunCluster [14], SAM-GS [4]。如果基因变化与表型存在相似性,那么FCS可以基于整体表达量
第三种:当想了解pathway的种类和其中各种联系的方向时
基因集的方法将pathway的联系视作无序排列,基因之间的关系也没有结构化,这样就丢弃了大量关于pathway描述的生物过程的信息,目前已经开发了一些基于拓扑的方法,想要在分析中加上这部分信息,也就是除了基因表达变化以外,还考虑每个pathway的所有基因的位置、作用以及互作信息。
第一种这样的方法是Impact Analysis[5],然后又陆续开发了30多种工具:us (Pathway-Express [5, 18], SPIA [27], ROntoTools [29], BLMA [22, 23]), as well as others (NetGSA [24], TopoGSA [9], TopologyGSA [20], DEGraph [16], PWEA [15], PathOlogist [11], GGEA [8], cepaORA, cepaGSA [12, 13], PathNet [6], etc.
第四种:想预测或解释下游/通路影响
基因集只是考虑某一pathway上的一组基因,并忽略了基因在通路上的位置,因此存在生物学解释的限制。如果某个通路由单个基因产物触发或通过单一受体激活,并且不产生特定的蛋白,那么这个通路可能受到很大的影响甚至完全关闭。
例如:胰岛素通路中不存在胰岛素受体( insulin receptor, INSR)【图中黄色的节点】,那么整个通路将会关闭(左图)。相反,如果几个基因参与到一个通路,但是只出现在下游的某个地方,那么它们的表达水平可能不会对这个通路产生那么严重的影响。【也就是说,了解基因所处的位置是很重要的】。如果使用基因集分析,那么它只能考诉你这组通路的基因是否在所有差异基因中富集,而不能告诉我们差异基因的变化是否会影响整个通路。
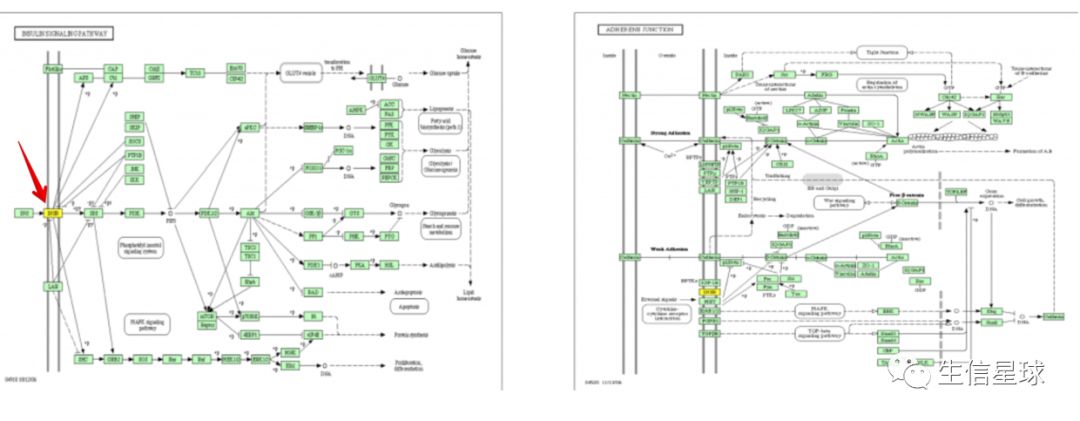
第五种:想寻找实验中明显被影响的机制
一些基因会具有多种功能或者参与到许多的通路,另外在每个通路中发挥的作用不同。例如,上面右图中显示的INSR(黄色节点)同样也是作为酪氨酸激酶受体蛋白参与Adherens Junction通路。如果INSR的表达发生改变,Adherens Junction通路可能并不会发生太大的影响,因为INSR仅仅是其中一个受体。
如果使用基因集,它不会考虑这些信息,如果不结合其他方法,仅仅用基因集分析时很难判断Adherens Junction通路或者胰岛素通路的变化幅度。
目前基因集虽然很全,但是其中各个通路中各种基因的互作关系还是没有被好好利用。而Pathway可以作为这一点的补充,因此如果想探究某一个特定的分子机制,首选还是pathway分析。
下面是iPathway的截图,其中对GSE47363数据集进行了pathway分析。实验利用了miRNA(miR-542-3p)处理细胞,想要理解这个miRNA的作用。利用 iPathway Guide 分析了处理组和对照组中表达量变化涉及的通路分析。图中红色的部分是自动推断了所有信号与不同基因的依赖关系得到的机制,从而做出的判断。而这个结果是不能从GSEA分析得到的。

第六种:想要结果结合最新的知识做出判断
随着数据量的增加,我们对各种通路的理解也在不断加深。因此可以根据不断更新的知识,在pathway图上增加、删除或者重新定向通路。而基因集是不能感知这种变化的,基因集能做的是:只要pathway中涉及相同的基因,即使它们之间相互作用随着我们研究的深入发生了改变,GSEA还是提供相同的结果。
如果只看上面6个特定,那么pathway分析好像更胜一筹,pathway具有更明确的生物学意义以及更准确的结果,但是为什么还要使用基因集富集分析(GSEA)呢?
想用GSEA第一种原因:想寻求更快的结果
GSEA的结果更加简单,因为不含有任何的拓扑结构信息,它们也更容易理解。通过计算富集的p值或者FCS打分(GSEA中提供的)就可以初步看下这一组基因是否可能与表型有关
想用GSEA第二种原因:当有自己定义的基因集
基因集分析不存在任何依赖关系 ,这也可能是一个优势。如果我们知道了一组基因可以在某一个通路中有协同作用,就可以快速将让它们定义为"基因集”,然后找到与表型可能的相关性。当然,其中可能会包含一些比较"随意”或者相关性不那么大的基因,就会妨碍对真正生物学通路的理解。
References
- Marit Ackermann and Korbinian Strimmer. A general modular framework for gene set enrichment analysis. BMC Bioinformatics, 10(1):1, 2009.
- William T. Barry, Andrew B. Nobel, and Fred Wright. Significance analysis of functional categories in gene expression studies: a structured permutation approach. Bioinformatics, 21(9):1943–1949, May 2005.
- Thomas Breslin, Patrik Eden, and Morten Krogh. Comparing functional annotation analyses with Catmap. BMC Bioinformatics, 5(1):193, 2004.
- Irina Dinu, John D Potter, Thomas Mueller, Qi Liu, Adeniyi J Adewale, Gian S Jhangri, Gunilla Einecke, Konrad S Famulski, Philip Halloran, and Yutaka Yasui. Improving gene set analysis of microarray data by SAM-GS. BMC Bioinformatics, 8(1):242, 2007.
- Sorin Draghici, Purvesh Khatri, Adi L Tarca, Kashyap Amin, Arina Done, Calin Voichita, Constantin Georgescu, and Roberto Romero. A systems biology approach for pathway level analysis.Genome Research, 17(10):1537–1545, 2007.
- Bhaskar Dutta, Anders Wallqvist, and Jaques Reifman. PathNet: A tool for pathway analysis using topological information. Source Code for Biology and Medicine,7(1):10, 2012.
- Bradley Efron and Robert Tibshirani. On testing the significance of sets of genes.The Annals of Applied Statistics, 1(1):107–129, 2007.
- Ludwig Geistlinger, Gergely Csaba, Robert Kuffner, Nicola Mulder, and Ralf Zimmer.From sets to graphs: towards a realistic enrichment analysis of transcriptomic systems. Bioinformatics, 27(13):i366–i373, 2011.
- Enrico Glaab, Anaıs Baudot, Natalio Krasnogor, and Alfonso Valencia. TopoGSA: network topological gene set analysis. Bioinformatics, 26(9):1271–1272, 2010.
- Jelle J. Goeman, Sara A. van deGeer,Floor deKort, and Hans C. vanHouwelingen. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics, 20(1):93–99, 2004.
- Greenblum, S. Efroni, C.Schaefer, and K. Buetow. The PathOlogist: an automated tool for pathway-centric analysis. BMC Bioinformatics, 12(1):133, 2011.
- Zuguang Gu, Jialin Liu, Kunming Cao, Junfeng Zhang, and Jin Wang. Centrality-based pathway enrichment: a systematic approach for finding significant pathways dominated by key genes.BMC systems biology, 6(1):56, 2012.
- Zuguang Gu and JinWang. Cepa: an R package for finding significant pathways weighted by multiple network centralities. Bioinformatics, 29(5):658–660, 2013.
- Corneliu Henegar, Raffaella Cancello, Sophie Rome, Hubert Vidal, Karine Clement, and Jean-Daniel Zucker. Clustering biological annotations and gene expression data to identify putatively co-regulated biological processes. Journal of bioinformatics and computational biology, 4(04):833–852, 2006.
- Jui-Hung Hung, Troy W Whitfield, Tun-Hsiang Yang, Zhenjun Hu, Zhiping Weng, and Charles DeLisi. Identification of functional modules that correlate with phenotypic difference: the influence of network topology.Genome Biology, 11(2):R23, 2010.
- Laurent Jacob, Pierre Neuvial, and Sandrine Dudoit. Gains inpower from structured two-sample tests of means on graphs. Arxiv preprint arXiv:1009.5173, 2010.
- Zhen Jiang and Robert Gentleman. Extensions to gene set enrichment. Bioinformatics, 23(3):306–313, 2007.
- Purvesh Khatri, Sorin Draghici, Adi L Tarca, Sonia S Hassan, and Roberto Romero. A system biology approach for the steady-state analysis of gene signaling networks. In CIARP’07 Proceedings of the 12th Iberoamerican conference on Progress in pattern recognition, image analysis and applications, pages32–41, Valparaiso, Chile, 13-16 November 2007. ACM.
- Sek Won Kong, William T Pu, and Peter J Park. A multivariate approach for integrating genome-wide expression data and biological knowledge. Bioinformatics, 22(19):2373–2380, 2006.
- Maria S Massa, Monica Chiogna, and Chiara Romualdi. Gene set analysis exploiting the topology of a pathway. BMC Systems Biology, 4(1):121, 2010.
- Cristina Mitrea, Zeinab Taghavi, Behzad Bokanizad, Samer Hanoudi, Rebecca Tagett, Michele Donato, Calin Voichita, and Sorin Draghici. Methods and approaches in the topology-based analysis of biological pathways. Frontiers in Physiology, 4:278, 2013.
- Tin Nguyen and Sorin Draghici. BLMA: A package for bi-level meta-analysis. Bioconductor, 2017. R package.
- Tin Nguyen, Rebecca Tagett, Michele Donato, Cristina Mitrea, and Sorin Draghici. A novel bi-level meta-analysis approach-applied to biological pathway analysis. Bioinformatics, 32(3):409–416, 2016.
- Ali Shojaie and George Michailidis. Analysis of Gene Sets Based on the Underlying Regulatory Net- work. Journal of Computational Biology,16(3):407–426, 2009.
- Aravind Subramanian, Pablo Tamayo, Vamsi K. Mootha, Sayan Mukherjee, Benjamin L. Ebert, Michael A. Gillette, Amanda Paulovich, Scott L. Pomeroy, Todd R. Golub, Eric S. Lander, and Jill P.Mesirov. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression. Proceeding of TheNational Academy of Sciences of the Unites States of America, 102(43):15545–15550, 2005.
- Adi L Tarca, Sorin Draghici, Gaurav Bhatti, and Roberto Romero. Down-weighting overlapping genes improves gene set analysis. BMC Bioinformatics, 13(1):136, 2012.
- Adi L Tarca, Sorin Draghici, Purvesh Khatri, Sonia S Hassan, Pooja Mittal, Jung-sun Kim, Chong Jai Kim, Juan Pedro Kusanovic, and Roberto Romero. A novel signaling pathway impact analysis. Bioinformatics, 25(1):75–82, 2009.
- Lu Tian, Steven A.Greenberg, Sek WonKong, Josiah Altschuler, Isaac S. Kohane, and Peter J. Park. Discovering statistically significant pathways in expression profiling studies. Proceedingof TheNational Academy of Sciences of the USA, 102(38):13544–13549, 2005.
- Calin Voichita, Michele Donato, and Sorin Draghici. Incorporating gene significance in the impact analysis of signaling pathways. In Machine Learning and Applications (ICMLA), 2012 11th International Conference on, volume1, pages126–131, Boca Raton, FL, USA, 12-15 December 2012.