014-懒惰也是一种美德
刘小泽写于18.7.19
所有的学习都是通过了解是什么、为什么后才有兴趣探究怎么做
Perl是什么?
Perl(Practical Extraction and Report Language)实用报表提取语言,1987年由Larry Wall设计,大写的P是语言本身,小写的p意为程序运行的解释器。
被称为“梦幻脚本语言”,Unix中的王牌工具,兼容了正则表达式,整合了awk、sed等一系列好用的小语法
他的优势在于:开源免费、跨平台、直接运行无需编译、无需定义各种变量及类型,不像其他语言上来就需要学习定义整型、浮点型等、perl很灵活,有单行模式,配合Linux十分好用
为什么用Perl处理生物数据?
Perl最大的优势就是处理字符串,而生信数据本质上就是将组织样本中的基因生物分子转为ATCGN这样的字符串来分析。
- Perl内置了强大的正则表达式,善于处理字符串【例如我们常用的操作:统计序列ATCG含量、基因组GC含量、核苷酸翻译成氨基酸、序列提取某个片段、碱基互补配对】像这样的任务,其他语言可能需要十几行甚至几十行,但是Perl有时只要一行!
- 处理生物数据,不可能只有一个样品吧。这么多的样品,如果你用在线工具,也需要一会的时间,但是Perl提供了批量化处理的操作,大大提高效率,本来一个小时的任务,你两分钟干完,剩下时间泡杯咖啡看集美剧都不为过!
- 许多软件的结果可能并不是我们现在想要的,例如blast比对大量的样本,结果往往还需要进行过滤才能符合我们的要求,Perl可以对其进行筛选并且提取出想要的序列;文件格式的转换也常常用到Perl;另外许多软件也就是用Perl编写的,会了Perl在看他们的源代码就容易多了,方便纠错
- Perl的作者就是语言学家,他开发的程序简单易懂
一个例子
数据结构
1 标量:
- Perl中最基本的数据类型
- 可为数字、字母,无需定义类型【其他语言需要提前定义类型:比如,整型还是浮点型,是单精度浮点型还是双精度浮点型】Perl中都是按照双精度浮点型进行保存和运算的,内部不存在整型。即使是整数也会被当成浮点数来计算,这样就省去了提前定义的麻烦
直接量:
程序中直接定义的一个值,例如-30,8e-3
数字操作:
常用的加减乘除、取模/取余数(%)【10%3=1】、乘幂3**2【=9】
字符串运算:
字符串是一连串的字符组合,字符可以是字母、数字、标点,可以为空,需要引号
生信数据比如处理序列就是处理字符串,例如给出“ACGCTAGCTAGCTCAGCTGGGCTAGCACGTCGATCGACTAGC” 计算他的长度、反向序列、互补配对、碱基替换、截取、翻译氨基酸【看!是不是就是那些所谓的网页小工具实现的内容?用Perl全能做到】
字符串中的引号差别:
- 单引号中的内容表示内容本身(但表示单引号和反斜线需要转义)
- 双引号支持反斜线转义,单引号不支持
字符串操作:
连接字符串:使用.连接两个文本,例如"hello"."world"
使用x 连接文本和数字,意思是重复几次,例如"fred"x3
【5x4 表示5555,5*4 表示20】
未定义的标量$_:
$_=undef 其他语言都要提前定义类型,Perl不需要提前定义,就等于先找个位置,用着什么就变成什么,比如用在数值计算中,他的值就是0;用在字符串中就表示空字符串; 或者刚读进来的每一行可以当成undef,一会再处理
#!/usr/bin/perl -w
$_="hello,bioinfoplanet";
print;
#结果就是hello,bioinfoplanet
2 标量变量
标量变量命名:
美元$ 表示,命令规范:
- 不能以数字开头
- 可以用字母下划线开头,后面可以加数字
- 区分大小写,尽量用小写
- 命令要能懂它的用途,勿求短
- 不要用内部保留的变量名
变量赋值:
单个等号是赋值【两个等号才是计算等于】
$dna="ATCACA"; 、$gc_sum+=3;
$dna.="ACTAG"; 得到的结果就是ACTAGACTAG
结尾以分号; 结束
3 数据比较
操作符优先级:
不需要记忆各个符号代表的优先次序,使用括号()可以自定顺序
比较操作符:
| 比较 | 数值 | 字符串 |
|---|---|---|
| 相等 | == | eq |
| 不等 | != | ne |
| 小于 | < | lt |
| 大于 | > | gt |
| 小于等于 | <= | le |
| 大于等于 | >= | ge |
4 换行符
了解这个非常重要!因为:有时在Linux下显示很好的文本文件移动到windows上用记事本打开后,所有的序列都会放在一行,十分的混乱,这是因为~
Linux中是换行
\n; Mac中是回车\r;Windows是回车加换行\r\n
Linux中使用cat A 显示出文件结尾的换行符:linux下结尾是$ ,mac下的结尾就是^M ,windows下就是^M$
因此,这也说明了windows下的文本文件到了Linux中文件大小会增加,就是因为每行相比linux多了^M
怎样解决这个问题?
dos2unix: windows转换为linux unix2dos:Linux转为windows unix2mac:linux转为mac mac2unix:mac转linux
chomp函数 只去掉结尾的换行符,没有换行符就不起作用
chop 函数 每次可以切掉结尾的一个字符,运行一次结尾少一个,既可以切换行符,又可以切回车符
5 列表与数组
列表(list)是指标量的有序集合;
数组(array)存储列表的变量
例如@array=[1,3,5,7,9]; 中@array就是一个数组,右边[1,3,5,7,9]就是一个列表
因此,打印的话,要么打印单个变量$,要么打印数组@
访问数组:
数组第一个值的下角标为0,$array[0]=1; $array[1]=3
统计数组中元素个数:
等于数组的最后一个角标$#array +1
【那么想要知道数组最后一个元素的值就可以用$array[$#array]或者$array[-1] 】
如何构建:
- 使用括号将元素括起来,并且之间逗号隔开;
- 使用范围操作符,适合连续加一的数值,例如
@number=(1..20)就是1-20这20个元素构成的数组; - 建立简单的字符串列表:如果每个字符串都加引号会很麻烦,可以这样:既不需要引号,也不需要逗号
@string=qw (i love bio info planet);【qw意思是quoted word】 另外括号作为界定符号,可以进行更换,将()换成!!//{}<>都可以
数组赋值:
($reed, $jessie, $ze)=("bio","info",undef);
print "$reed\n";
print "$jessie\n";
print "$ze\n";
得到结果:
bio
info
【第三行为空】
刘小泽写于18.7.20~练习了一段时间,发现最大的疏忽就是结尾的
;切记
两个数组相关函数:split和join【也就是标量和数组间的转化】
split: 将字符串按照固定的分隔符进行切割,切完后得到一个数组 【后接三部分:分隔符,放在两个斜线之间;要分隔的字符串;分隔的份数,可以不写】
#!/usr/bin/perl -w
$test="bio;info;planet";
@array=split /:/, $test;
print "@array\n"
# 结果是
# bio info planet【注意并不是输出一个换一行哦,而是整个数组在一起输出后再换行】
join:与split相反,将数组连接成一个标量
【后面冒号中的内容为分隔符,这里除了:也可以用制表符\t,它等于四个空格,而用制表符分隔的一个好处就是:用excel打开后会识别这几项内容并显示在不同的列中】
$test2=join ":", @array;
print "$test2\n";
对数组进行操作:
对数组尾部操作
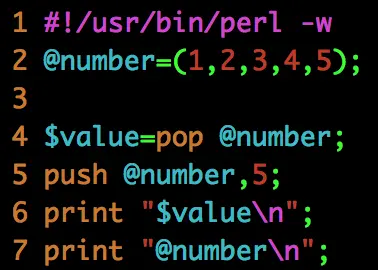
pop:弹出数组中最后一个元素
**push:**向数组中最后一个位置加入一个元素【常用】
对数组开头操作 shift : 取出开头第一个元素【常用】 unshift:在开头第一个位置加入一个元素
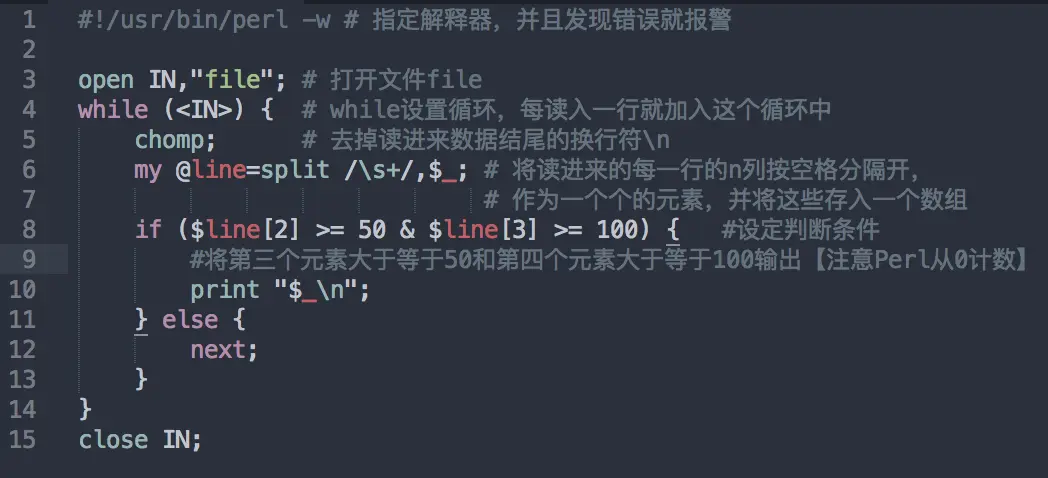
#用Perl处理一个矩阵时,第一列往往是ID信息,可以提取出来方便后来排序 while (<IN>) { chomp; my @line=split /\s+/,$_; #\s匹配任意空白符,包括空格、制表符,+表示重复之前一次或多次 my $id=shift @line; #my限定变量在当前循环体或函数体中 print "$id\n"; }对数组进行排序 sort:默认按照ASCII码由大到小排序,比如排序1-10,他会把10排在1的后面2的前面 reverse :逆向排序,不仅可以对数值还能对字符进行排序。例如:找到一段DNA序列的反向序列
#!/usr/bin/perl -w $dna="ACGCTACGATCGACAGCATCGACTA"; $dna_rev=reverse $dna; print "$dna\n" print "$dna_rev\n"遍历数组
#!/usr/bin/perl -w @number=(1..6); # foreach格式 foreach $num (@number){ print "$_\n"; }
6 上下文
同一个表达式,出现在不同的地方会有不同的意义【Perl中总是根据上下文来返回对应的值,主要看对标量进行操作还是对数组进行操作】
为什么要时刻关注上下文呢?
#!/usr/bin/perl -w
@name=qw(bio info planet);
@sorted=sort(@name);
print "@sorted\n"; # 结果返回 bio info planet
$num=10+@name;
print "$num\n"; # 结果返回13
这个例子告诉我们,在Perl这个灵活多变的大环境中,同样的表达式@name用在不同地方,他都是对的。再一次印证了Perl的哲学思想:解决问题的办法不止一个!
7 获得帮助
perldoc 获得内置帮助
-q xxx 在perldoc帮助文档中寻找包含xxx关键词的文档 -f 搜索Perl内置函数的功能 -v 搜索内置变量,如 perldoc -v @ARGV
perldoc perlfaq 查看关于Perl的经常被问到的问题及答案 perldoc perlop 查看Perl的操作符及优先级 perldoc perlsyn 查看Perl的的语法结构 perldoc perldiag 查看警告和错误相关的内容 perldoc perlvar 查看与变量相关的内容
内容读写
1 输入输出
使用open函数
#比如进行dna向蛋白质序列的转换
#!/usr/bin/perl -w
use strict;
open IN, "<dna.fa";
open OU, ">protein.fa";
输入输出整个过程有五部分:
open函数打开文件
像IN/OU这种叫做文件句柄(Perl中处理进程与用户交互的名称,IN说明用户让Perl输入某个文件) 【关于文件句柄:命名由字母、数字或者下划线组成,不能以数字开头,通常都是用大些字母命名;内置6个,STDIN、STDOUT、STDERR、DATA、ARGV、ARGVOUT】
逗号分隔【不能省略!】
引号部分是文件路径和名称
<、>是输入、输出方向【一旦输入文件用了> 就会被清空】;而>>是追加,如果原文件不存在,就会创建一个新文件【和Linux重定向方式是一样的】文件路径
上面👆的操作中,如果只有一个文件还好,如果有多个文件,那岂不是每次要修改
open IN/out的参数?
因此使用@ARGV就十分重要了!
@ARGV存储的是命令行的参数 ,例如:
#命令行运行 perl dna2pro.pl gene.fa pro.fa ref
# 其中dna2pro.pl是$0; gene.fa是命令行第一个参数,在perl语句中存储为$ARGV[0]; 以此类推,pro.fa是$ARGV[1]; ref是$ARGV[2]
2 读取数据:主要靠句柄
#假设命名为test.pl
#!/usr/bin/perl -w
open IN, "<$ARGV[0]";
$first=<IN>; #<>是钻石符,表示既可以是输入可以是输出,就把IN放在数据流符号的中间
$second=<IN>;
$third=<IN>;
print "$first\n$second\n$third\n";
命令行中输入perl test.pl dna.fa结果就得到前三行内容
但是这样一个一个定义有些麻烦,尤其行数比较多的时候,因此可以用while
#!/usr/bin/perl -w
open IN,"@ARGV[0]";
open OU,"@ARGV[1]";
while (<IN>){
chomp;
print OU "$_\n" #每行读入的内容存储在了$_中
}
close IN; #close关闭文件句柄
close OU;
命令行中输入perl test.pl in.fa out.fa 将读取行内容直接输入到out.fa中,不会打印到屏幕上
总结一下读写基本操作: 首先使用open函数打开文件,然后while循环和句柄读入文件,并使用换行符,进行处理后将结果输出,最后关闭句柄
3 格式转换
将fq转为fa:
区别两种格式:
fq格式有四行,fa文件只有两行(即不需要fq的第三行和第四行);
fq开头以@开头,fa文件开头是 >
#!/usr/bin/perl -w
open IN,"zcat $ARGV[0] | "; #针对压缩格式的fq文件,也不需要预先解压缩
open OU,">$ARGV[1]";
while ($id=<IN>) { #这样就将第一行命名为了id
chomp ($id); #对读进来的第一行去换行符
chomp ($seq=<IN>); #对读进来的第二行(命名为序列行)去换行符
<IN>;
<IN>; #第三、四行用不着,所以不需要操作
$id=~ tr /@/>/; # 替换第一行的@为>,使用tr函数
print OU "$id\n";
print OU "$seq\n";
}
close IN;
close OU;
写于18.7.22
哈希
哈希数据是没有顺序的;使用键值对,键是唯一的,值可以相同;使用大括号调取键【数组是用中括号,这个区别要注意】;引用哈希变量用
%【标量变量用$,数组变量用@】;哈希中存储的键值对形式:键=>值
定义空哈希:
%hash=();
将列表改成哈希:
例如有这么一份表格,表示字母与数字的对应:
| wordA | one |
|---|---|
| wordB | two |
| wordC | three |
| wordD | four |
| wordE | five |
#!/usr/bin/perl -w
use Data::Dumper; #这个模块可打印出哈希的数据结构
%hash=();#先定义一个空的哈希,一会向其中存入内容
open IN,"<$ARGV[0]"; #文件重定向输入
while (<IN>){ #一行一行读入该文件
chomp; # 每一行例如wordA one 去掉结尾的换行符
@line=split /\s+/,$_; #按照空格分隔,将两列存储在line数组中【$_是split的作用对象,也就是读入的每一行】
$hash{$line[0]}=$line[1];#将第一列作为键,第二列作为值存储在之前的空哈希中。
}
close IN;
print Dumper (\%hash); #print加Dumper模块,再引用哈希
对哈希进行遍历:
keys函数
# keys函数也常常用在对哈希的遍历过程中
#keys和value函数定义键和值,保存到数组并打印
@key=keys %hash;
@value=value %hash;
print "@key\n";
print "@value\n"
#返回的键值对还是一一对应的,但是键值对之间的顺序和原来列表不同
#如果将键用$存到标量而不是用@存到数组,那么返回的就是键的个数
#$key=keys %hash; 结果返回的就不是具体的键wordA-E,而是haxi的元素个数5
- 对哈希进行遍历也可以用
foreach
# 实际上foreach是用来对数组进行遍历的,这里的@tmp就是用keys对哈希进行遍历再排序得到的数组
@tmp=sort keys %hash;
foreach (@tmp){
print"$_\n"
print "$hash($_)\n;"
}
while循环中用each
while (($key,$value)=each %hash){
print "$key\n$value\n";
}
检测哈希中某个键是否存在:
if (exists %hash{wordA}) {
print "$hash{wordA}\n";
}
用哈希进行序列提取:
根据固定的id从另一个文件或者数据库提取出序列
一般处理方式是:将小的数据集存储到一个哈希中,然后再遍历大的数据集,每次判断id在哈希中是否存在,如果存在就输出,不存在继续循环
就想NCBI blast过程,就是根据id去找序列的过程。【那么如果将大的数据集存到哈希中,用小数据集遍历可以不可以实现呢?其实也可以,但是会消耗大量的资源,不值得!就想去blast,如果将大的NCBI nt数据库存到哈希,那么这个数据库有80多个G,是很慢的】
# 第一步:将gene id【第一个参数】存储到哈希中
#!/usr/bin/perl -w
use strict;
use Data::Dumper;
if (scalar @ARGV==0){ #添加帮助信息,如果ARGV这个命令行参数个数为0,即没有输入的id文件和基因序列文件
die "Usage: This program is used to extract sequence by a gene id from a gene list
perl $0 <id list> <fasta file>";
} #die函数终止程序,并打印返回信息;$0意思是程序的名字
my %hash=();
open IN,"<$ARGV[0]";
while (<IN>) {
chomp;
$hash{$_}=1;
}
close IN;
#print Dumper (\%hash);
# 第二步:读取基因序列文件【格式是每个id对应一条序列】
open FA,"<$ARGV[1]"; # 打开fasta格式序列文件
$/=">";<FA>; #根据fasta的格式,将分隔符$/定义为>,这样每次读入的数据就是一个字段,也就是一个完整的基因序列。包括了id和序列
while (<FA>) { # 每次读入一个分隔后的基因
chomp;
my id=(split /\n/$_,2)[0]; #将id与序列分开~利用换行符进行分隔,存为一个为定义标量。最后加上的数字2,意思是将标量分成两份,也就是id行和序列行。然后就可以根据基因id,即分隔后的[0]进行判断
if (exists $hash{$id}){ #判断第一步生成的哈希中的值在id中是否存在
print ">$_"; #因为原来gene id是带>的,但是$id中用$/=">"处理后就不带>,所以最后输出还需要加上>;另外输出的结果是$_,即id+序列
} else {
next; #一个一个判断,如果不存在就检查下一个
}
print "$id\n"
}
close FA;
写完脚本可以用
perl -c查看脚本是否正确
子程序
之前用过了chomp、print等,perl还能让用户自定义函数,方便利用已有的代码
使用sub定义【不能以数字开头,最好名字和函数功能相关,不要和内置函数重名】
每个子程序都有一个返回值,而运行子程序要的就是返回值
最简单的使用:
#以打印生信星球信息为例
#!/usr/bin/perl -w
use strict; #代码中有不好的编码风格,那么提示编译失败
&print_planet; # &符号可以调用子程序【定义的和系统不冲突时可以省略&,冲突时必须加上&】
sub print_planet {
print "Hello, bioinfoplanet\n";
}
&print_planet;
#这样结果就会输出两次"Hello, bioinfoplanet"
写于18.7.23
子程序的参数与返回值–输入与输出:
上面的简单使用,没有加上任何参数就会输出字符串这样一个标量;
当然,也可以对子程序输入数组、哈希、文件等参数,然后输出标量、数组或者哈希。
默认将子程序的最后一个表达式的结果作为子程序的返回值,如果想返回中间一个表达式,使用 return 函数
这里以一个加法运算来学习:
#!/usr/bin/perl -w
use strict;
sub sum {
my $num1=shift @_; #定义第一个标量变量为@_中取出的第一的第一个元素
my $num2=shift @_;
my $total=$num1+$num2;
return $total;
}
my $total=&sum (3,5);# &sum()括号中的标量会存储到@_这个数组变量中;括号中有几个参数,@_数组中就有几个元素
print "$total\n"
统计fasta信息的子程序:
程序思路:输入有一个,gene id与序列文件对应的哈希列表【格式为id=序列文件】。输出要有三列,一列是gene id,一列是基因数量,一列是基因平均长度。
读取fasta文件时,将$_修改分隔符为>,这样一次就能读进来整段序列,将每段序列存储到哈希中,序列的id为哈希的键,序列为值
#!/usr/bin/perl -w
use strict;
open IN,"<$ARGV[0]"; #输入的第一个文件是基因id与序列对应的哈希文件
while <IN> {
chomp;
my ($id, $file)=split( /=/,$_)[0,1]; #哈希文件的格式是id=序列文件,因此用=分隔二者,存到$_,再将他的第一列赋给变量$id,第二列赋给变量$file
my ($gene_num,$gene_len)=&gene_stat ($file) #定义基因数量、长度的变量,子程序是gene_stat,对上面得到的$file也就是序列文件进行操作
print "$id\t$gene_num\t$gene_len\n";
}
close IN; #while循环是主体,其中包括了子程序
sub gene_stat { #开始定义子程序
my $file=shift @_; #先定义几个变量:file就是刚才得到的数组变量@_中元素,定义基因数量、长度初始值为0
my $gene_num=0;
my $gene_len=0;
open FA,"$file"; #将读入的$file序列文件作为FA句柄
while (<FA>){
chomp;
if (/^>/) { #判断读入的文件开头为>,则作为基因数目
$gene_num+=1;
} else {
my $len=length ($_); #否则作为基因序列,统计长度,一会计算平均长度用
$gene_len+=$len
}
}
close FA;
my $avg_len=$gene_len/$gene_num;
return $gene_num, $avg_len;
}
Perl常见问题:
Perl的编程过程中,会出现各种各样的问题,需要不断调试。有时候很头疼的就是,即使按照别人的程序敲一遍还是会报错
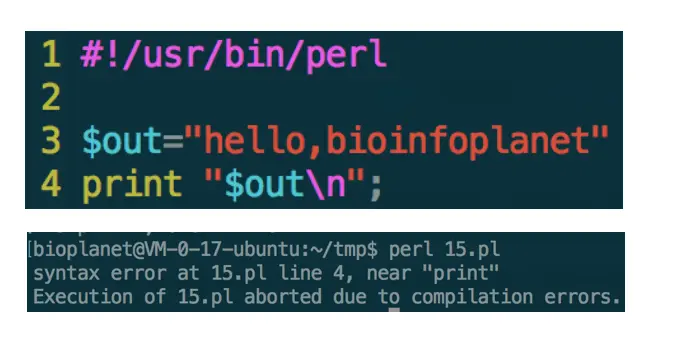
错误一:忘记加分号
分号在Perl中代表一个完整的语句,忘记加分号就会提示语法错误,并且运行会给出具体出错行数
这里显示第四行有问题,如果在一个大程序中,要快速跳转到某一行,使用vi +4 1.pl ,因为print上一行没有分号
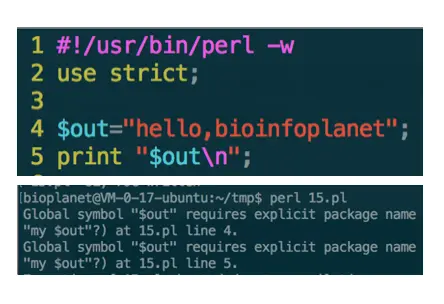
错误二:是否需要用my定义
很多时候需要使用use strict 编译命令,这是就要要使用my 来编译变量,而且不能重复定义
#使用strict的例子:
#!/usr/bin/perl -w
use strict;
my $total;
my $num1=2;
my $num2=1;
$total=$num1+$num2;#这里的total因为之前定义过了,所以不用再使用$total
print "$total\n";
错误三:拼写错误
这个在很多编程中都会存在,拼写错误会提示变量没有被声明
这个问题在文本编辑器如Vim中会帮助我们解决,如果某个变量输入错误,比如print输入为prnit ,Vim会通过颜色不同(正确有颜色,错误为白色)
对于变量输入,最好使用vim自带的ctrl+N 或ctrl + P 的自动补全功能,这样可以避免错误
错误四:大括号的使用
尤其使用if嵌套循环时,每一个大括号是一个程序块,如果缺失某一对配对的大括号其中一个,就会提示Missing right/left curly or square bracket at line xxx ;如果大括号多了一个,会提示Unmatched right/left curly bracket at line xxx
避免报错小技能:
perl -c(check)运行之前先检查一下是否书写有问题perl- d(debug)一种交互的perl编程模式,适用于长的脚本。l列出命令的10行;如果要列出某几行,就用l 4-7这样的l+行号详细信息可以参考perldoc perldebug
正则表达式:重要的特性!
Perl的正则表达式是内建的,为Perl提供了快速、灵活的字符串处理
Perl中的正则也叫做“模式匹配”,是用来匹配某字符串的模版; 日常使用中,模式匹配无处不在,例如使用搜索引擎,在搜索框内输入关键字,就能返回信息,他就是利用了正则表达式
一个简单的例子:
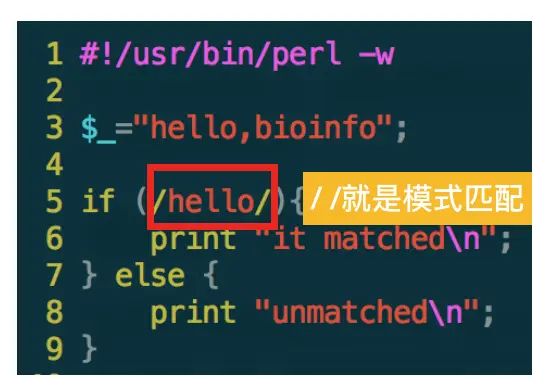
元字符:
.替换单个字符(一对一替换),不能替换换行符\转义符,例如匹配.或者\本身要使用\表示数量的
* + ?*【例如在root用户下,使用rm -rf *叫做“自杀模式”】,与.配合使用表示任意字符出现0次或多次 【0—∞】+表示至少一次 【1—∞】?表示0次或1次【范围最小】花括号中指定重复次数,例如匹配1-5次bioinfo中的o
bio{1,5}info如果要制定整个单词次数,用小括号进行分组
/(bioinfo){3}/这个表示匹配bioinfo3次;/(bioinfo)+/表示匹配bioinfo一次以上
关于字符集:
- 匹配字符集的单个字符
/[bcdefwz]at/匹配bat、cat、fat其中一个,等同于/(b|c|d|e|f|w|z)at/或者更简单的/[b-fwz]at/(表示b-f连续的字母加wz) - [a-z]、[A-Z]、
[0-9]简写为\d+、[a-zA-Z0-9_]也就是单词字符,简写为\w \s+表示空白,例如split /\s+/,$_;- 反义字符集:利用
^,例如[^0-9]等于[^\d]等于\D,表示不匹配数字 【反斜线加大些字母表示不匹配】
- 匹配字符集的单个字符
要排除某个匹配,用
!
#来吧!请上手操作一下吧!
#打开你的Linux terminal,新建一个perl脚本
#!/usr/bin/perl -w
use strict;
print "Do you like bioinfoplanet?\n"
my $choice=<STDIN>;
if ($choice =~ /y e s/ixs) { #匹配的时候用 =~ 加模式/ /
print "Thanks, good job!\n"
} else {
print "OK, we'll do better!\n"
}
# 介绍一下$choice =~ /y e s/ixs中的修饰符
# i忽略yes其中各个字母的大小写;
# x可以忽略模式中的空白【常常针对模式较长,都是点号、星号、字符集等,这样写在一起就看着很复杂,需要加空格隔开,但是运行程序的时候又想让他们连在一起,所以用x】
# s可以让点号匹配到换行符,这样即使 "Do you like bioinfoplanet?"这一句是分行显示的,也可以匹配成功
# m:多行匹配,跨行模式匹配
# g全局匹配
生信模式匹配小练习:
现在有一些序列,下载地址https://github.com/Bioinfoplanet/Perl-Test.git 打开后是这样:
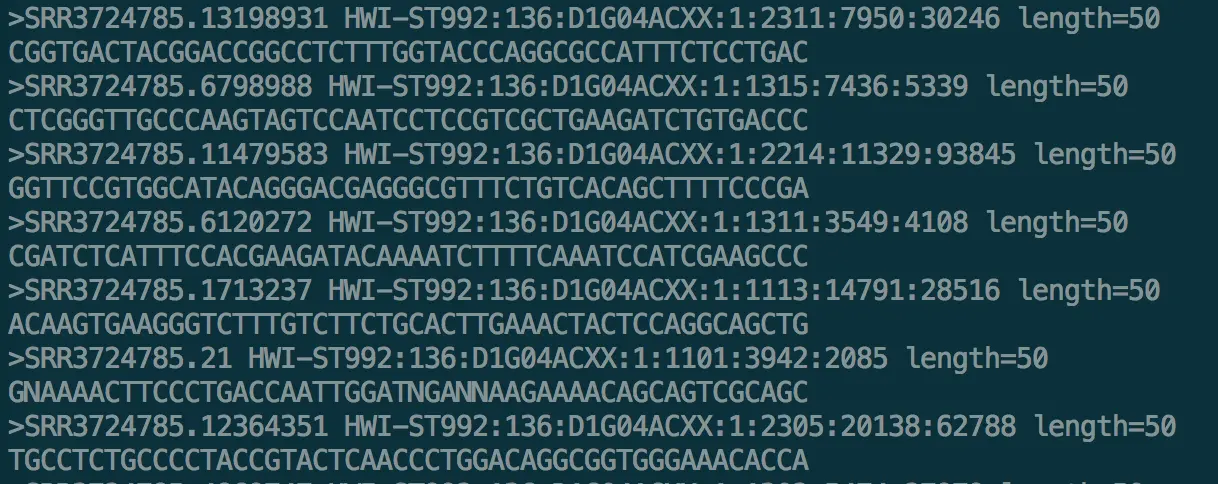
想寻找开头是CCG,结尾是TTG的序列
#!/usr/bin/perl -w
use strict;
open IN,"<$ARGV[]0";
while (<IN>) {
chomp;
if (/^CGG.*TTG$/i) { #^表示开头匹配,$表示结尾匹配
print "$_\n";
} else {
next;
}
}
close IN;
#补充一个vim小技巧,利用d(删除)再结合^或$,可以表示删除光标到行首/尾
单词锚定:
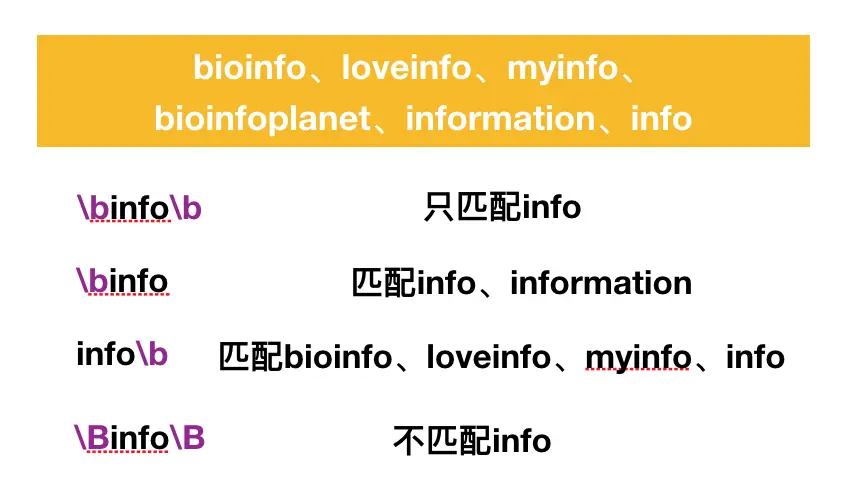
挖掘文本信息:
一般本地blast的format 0结果中,会有匹配的分值(Score)、期望值(Expect)、一致性(Identities)、准确度(Positives)、Gaps等信息,可以挖掘这些信息
#比如这里有一个blast format0的结果,result1中想要数字部分,result2想要括号中的百分数部分
#!/usr/bin/perl -w
use strict;
my $result1="Score = 150 bits (308), Expect = 1e-34, Method: Compositional matrix adjust.";
if ($result1=~ m/Score\s*=\s*(\d+)\s*(?:bits|Bits)\s*\(\d+\)\,\s*Expect\s*=\s*(\S+),\.*/) { #匹配过程看起来很复杂,其实也就是一点点填充的过程,有空格就用\s*,有数字就用\d+,对于中间没有空格的整体如1e-34就直接使用\S+。就像这样把整个句子用元字符的方式重绘出来,方便大量统计
print "$1\t$2\n"; #$1与$2就是捕获之前分组的变量
}
#得到的结果就是 150 1e-34
#按说每一个括号就是一个分组,而后面用$来捕获变量也是根据括号,那么比如(bits|Bits),我们只想让他用来分组,表示匹配bit或者Bits。但是这样有一个问题,后来引用的时候就会讲这一部分也输出;为了避免这个问题,就在左边括号后面加上?:表示这个整体就是一个分组,别把它当变量使用
my $result2="Identities = 131/468 (28%), Posotives = 217/468 (46%), Gaps = 46/468 (10%)";
if ($result2=~ m/(\S+)/g) {
print "$result2\n";
}
改进版:
#!/usr/bin/perl -w
use strict;
my $result1="Score = 150 bits (308), Expect = 1e-34, Method: Compositional matrix adjust.";
my $result2="Identities = 131/468 (28%), Posotives = 217/468 (46%), Gaps = 46/468 (10%)";
my @info1= ($result1=~ m/Score\s*=\s*(\d+)\s*(?:bits|Bits)\s*\(\d+\)\,\s*Expect\s*=\s*(\S+),\.*/);
print "@info1\n";
#直接将捕获的变量存储到列表中,即使没有匹配到,也只是会输出一个空列表
my %info2= ($result2=~ m/(S+)/g); #加上g意思是将全部符合条件的找出来
print "@info2\n";
另外还有一个s(替换模式),例如
#!/usr/bin/perl -w
use strict;
my $line="hello, bioinfoplanet";
$line=~ s/hello/nihao/;#结果就将hello变成了nihao
$line=~ s/^/Hi,/; #结果为Hi,nihao, bioinfoplanet
$line=~ s/$/hi/; #结果在结尾加上一个hi
$line=~ s/hello//g; #将全部hello替换为空,大规模替换要加上g
print "$line\n";
对序列进行处理:
得到序列的反向/互补序列
#!/usr/bin/perl -w
use strict;
my $seq="TTCGATGCTAGCATCGATCGCTAGCATCGCTAGCATCCCGATTGACTAATCAT";
my $qes=reverse $seq;
print "$qes\n";#序列反转
$qes=~ tr/ATCGN/TAGCN/; # 序列碱基互补
print "$qes\n";
#结果1得到:TACTAATCAGTTAGCCCTACGATCGCTACGATCGCTAGCTACGATCGTAGCTT
#结果2得到:ATGATTAGTCAATCGGGATGCTAGCGATGCTAGCGATCGATGCTAGCATCGAA
序列全变大些/小写
#!/usr/bin/perl -w
use strict;
my $seq="TTCGATGCTAGCATCGATCGCTAGCATCGCTAGCATCCCGATTGACTAATCAT";
$seq =~ s/(\w+)/\L$1/g; #(\w+)是将所有字母分成一组,\L是小写[\U是大写],$1是捕获前面的分组【另外\u是更改首个字母大写;\l首字母变小写】
print "$seq\n";
#结果得到:ttcgatgctagcatcgatcgctagcatcgctagcatcccgattgactaatcat
另外这些大小写语句除了在s模式下使用,还能在双引号内使用,例如在print 语句中格式化输出
print "\Uhello, wor\Eld" 输出结果是HELLO, WORld 【注意这里的\E是终止前面\U命令的意思】
贪婪匹配 – 匹配尽可能多的内容
一般. + ? 会匹配尽量多的内容,比如上个例子$seq 中,如果匹配模式是$seq=~ /^TTC(.+)CAT/i ,即使中间也出现了CAT,程序也不会停下,他会一直匹配到结尾的CAT,这就是贪婪;
贪婪的对立面就是节俭,这种模式可以在. + ? 的基础上加上? ,也就是$seq=~ /^TTC(.+?)CAT/i 就输出最短的序列
格式化序列
有时一个fasta文件中序列长短不一,并且有的换行有的不换行,这样看起来就很难看。这时就可以将序列格式化,让每行保证一定量的字符【可以自定义】,并且加换行符。
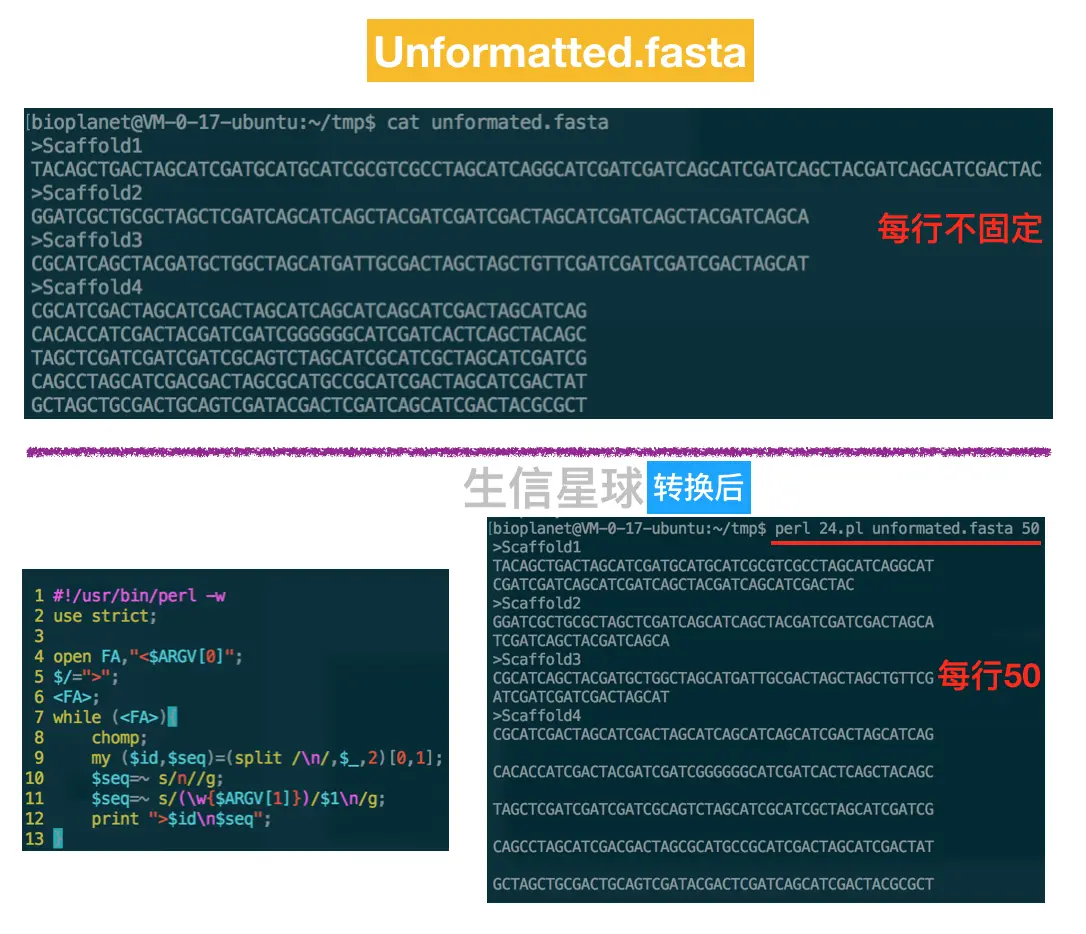
测试fasta下载:
wget https://github.com/Bioinfoplanet/Perl-Test/blob/master/unformated.fasta
#!/usr/bin/perl -w
use strict;
open FA,"<$ARGV[0]";
$/=">";
<FA>;
while (<FA>) {
chomp;
my ($id,$seq)=(split /\n/,$_,2)[0,1];
$seq=~ s/\n//g;
$seq=~ s/(\w{$ARGV[1]})/$1\n/g; #每x个字符作为一个$1,也就是前面选取的(\w{})得到的内容,注意\w后面是大括号而不是小括号
print ">$id\n$seq"
}
写于18.7.25
数据加千分位
一大长串数字如果加上千分位将会很容易观察,千分位只在整数部分添加(从后往前),小数部分不添加
#!/bin/bash/perl -w
use strict;
my $num=123456789.98;
1 while $num=~ s/^(-?\d+)(\d\d\d)/$1,$2/; #建立一个死循环,匹配模式:正或负(-?)数(\d+),后面的\d\d\d是从后向前每三个数划分一次,每次循环的结果即每三个数字用逗号隔开
print "$num\n";
#结果就是123,456,789.98
或者可以通过定义子程序,之后就可以直接用子程序来添加
#!/usr/bin/perl -w
use strict;
my $num=123456789.98;
my $result=&delimiter ($num);
print "$result\n";
sub delimiter {
my $data=shift @_;
1 while $data=~ s/^(-?\d+)(\d\d\d)/$1,$2/;
return "$data\n"
}
- 输出特定长度的序列
有时需要对fa序列提取部分序列进行比对,提取的起始位置、终止位置以及输出的每行长度都可以定义

基因组信息统计:
#!/usr/bin/perl -w
use strict;
#先定义全局变量,也就是从赋值到输出结果一直有效的变量
my $total_len=0;
my $total_num=0;
my $total_gap=0;
my $total_gc=0;
my @len_array=(); #排序之后取出首尾元素,也就是最短和最长序列
print "ID\tGeneLength(bp)\tGapLength\tGCcontent(%)\n"; #添加表头
open IN,"<$ARGV[0]", or die "can not open this file$!\n";
$/=">";<IN>; # $/代表默认的输入记录的分隔符,默认为\n,这里指定为>
while (<IN>) {
next if (/^\s+$/); # 如果行为空则不处理
chomp; # 去除$/(也就是默认的\n)
my ($id,$seq)=(split /\n/,$_,2)[0,1]; # 将id与seq行分开
$seq=~ s/\n//g; #对seq行进行统计前,要把seq中的换行符去掉
my $len=length ($seq);
push @len_array,$len; #向数组最后一个位置存入
$total_len+=$len;
my $gc+=($seq =~ s/G/G/g + $seq =~ s/C/C/g); #通过全局替换获得G、C字符的个数
my $GC=($gc/$len)*100; #gc含量等于G和C个数之和除以每条序列的长度
my $gap+=($seq =~ s/N/N/g);
$total_gap+=$gap #将计算的gap数加入全局变量中
$total_gc+=$GC;
print "$id\t$len\t$gap\t";
printf "%.2f\n","$GC"; #格式化输出,让GC结果保留小数点后两位
$total_num++; #代表一次循环结束
}
my $avg_len=($total_len/$total_num);
my $total_GC=($total_gc/$total_len)*100;
my @sort_len=sort {$a<=>$b} @len_array; #将数组中序列长度排序,方便下面输出最长最短序列
print "\nTotal Stat:\n";
print "Total Number {#}:$total_num\n";
print "Total Length (bp):$total_len\n";
print "Gap(N)(bp):$total_gap\n";
print "Average length(bp):%d\n", "$avg_len";#整数输出
print "Minimum Length(bp):$sort_length[0]\n";
print "Maximum Length(bp):$sort_length[-1]\n";
printf "GC content(%):%.2f\n","$total_GC";