013-VCF格式
刘小泽写于18.7.17 所有的数据,一旦要找变异位点信息,就离不开VCF。豆豆也是在写一个重测序的操作流程,遇到了VCF文本,之前也是没有了解过,这次再多学一个格式 官方最新说明文档https://samtools.github.io/hts-specs/VCFv4.3.pdf 这个文件格式的解读会比以往各种文件都要重要,有了它就能直接进行分析了,属于离下游可视化最近的数据
VCF是什么?
这个东东的全名是:Variant Call Format【真的是“简称让我无地自容,全称使我无师自通啊”】 它是存储变异位点的标准格式,可以用来表示单核苷酸多态性(SNP)【在人类基因组中,分布普遍并且密度比较大,总数超过10^7^, 平均每300bp就有一个SNP 】、插入缺失(InDel,也就是短片段的插入与缺失)、结构变异(SV: Structural Variant,也就是大片段的插入与缺失) 、拷贝数量变异(CNV:Copy Number Variant)【**说一下CNV:**比如一个基因在染色体的一条染色单体上的数目为1,但是在染色体复制过程中,不知为何,复制结束后该基因在染色单体数目由1变成了2或者n。尤其在人类基因组中存在大量大于1 kb但小于3 Mb的DNA片段多态。它发生的频率远远高于染色体结构变异,并且整个基因组中覆盖的核苷酸总数大大超过SNP的总数】
变异的生物学基础
遗传与变异:遗传就是将亲代的遗传物质传递给子代,这样保持了世代交替中的稳定性;但是,这种稳定性又非一成不变,每个个体的基因组随着时间和空间的变化,导致结构发生改变,从而产生了不同于亲本的性状,这就是变异。总而言之,遗传保持了物种代代之间可持续性,变异提供了长期进化过程中的适应性,又促进了进化。
同一个亲本的子代在不同时间、不同空间与环境相互作用,就会出现不同的基因型和表型,基因检测就是通过检测不同样本基因组上的差异,来推断基因型与表型之间的关系,还有与环境之间的互作关系。
基因组上的变异主要有单个碱基的变异(转换、颠换、插入、缺失)以及染色体水平的变异(插入、缺失、易位、倒位)。这些表现在基因组检测上就是:SNP、InDel、SV、Corpangene(插入缺失发生在基因水平)、CNV(发生在串联重复区的插入与缺失)
变异物质基础:
基因组发生变异的原因主要可以分为自发突变和诱发突变。之所以能够累计这么多突变,主要由于自发突变。而引起自发突变的原因也有很多,比如DNA复制过程中由于DNA聚合酶产生错误、DNA物理损伤、转座等,但这些错误和损伤大多会被自身的修复系统修复,那么为什么还会有这么多变异位点存在呢?
这是因为~
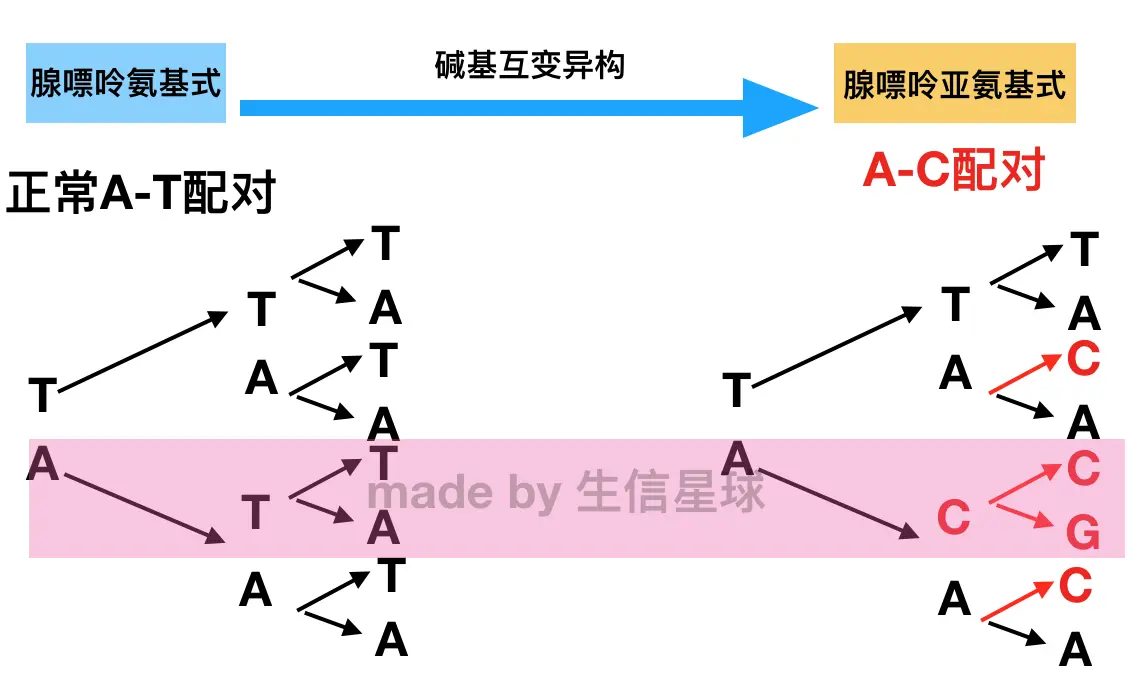
碱基能够以互变异构体的不同形式存在【A-T配对变为GC的过程】
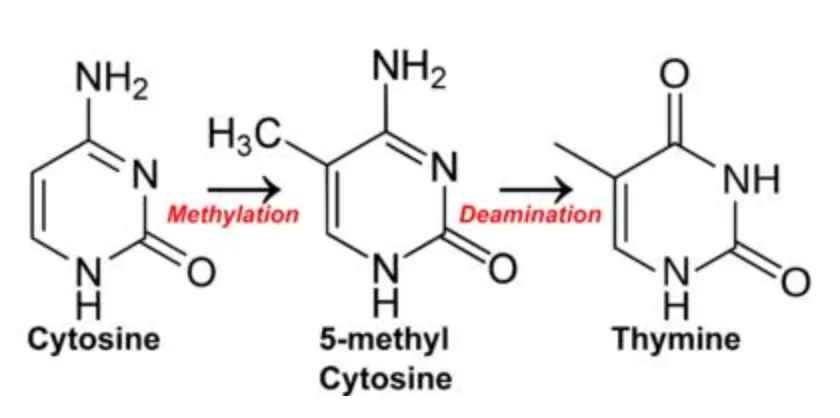
InDel:碱基有时会从核苷酸移除,留下一个叫做脱嘌呤或者脱嘧啶的缺口,进行下一轮复制时不能够正常配对。【原因可能是胞嘧啶自然脱氨基形成尿嘧啶,因为尿嘧啶并非DNA的碱基所以被DNA自身的修复系统识别并清除,留下一个空位】
转座子:随机插入到基因组,就相当于基因组上一个位点被复制/剪切后粘贴。当有多个这种序列结构时,他们之间就是同源的,因此就会导致同源重组【同源重组:非姐妹染色单体(sister chromatin) 之间或同一染色体上含有同源序列的DNA分子之间或分子之内的重新组合,进而引起缺失、重复、倒位等】
VCF文件怎么用?
VCF使用UTF-8编码,有两大部分:一部分是注释信息(以##开头),一部分是具体突变信息 【给出两个例子】
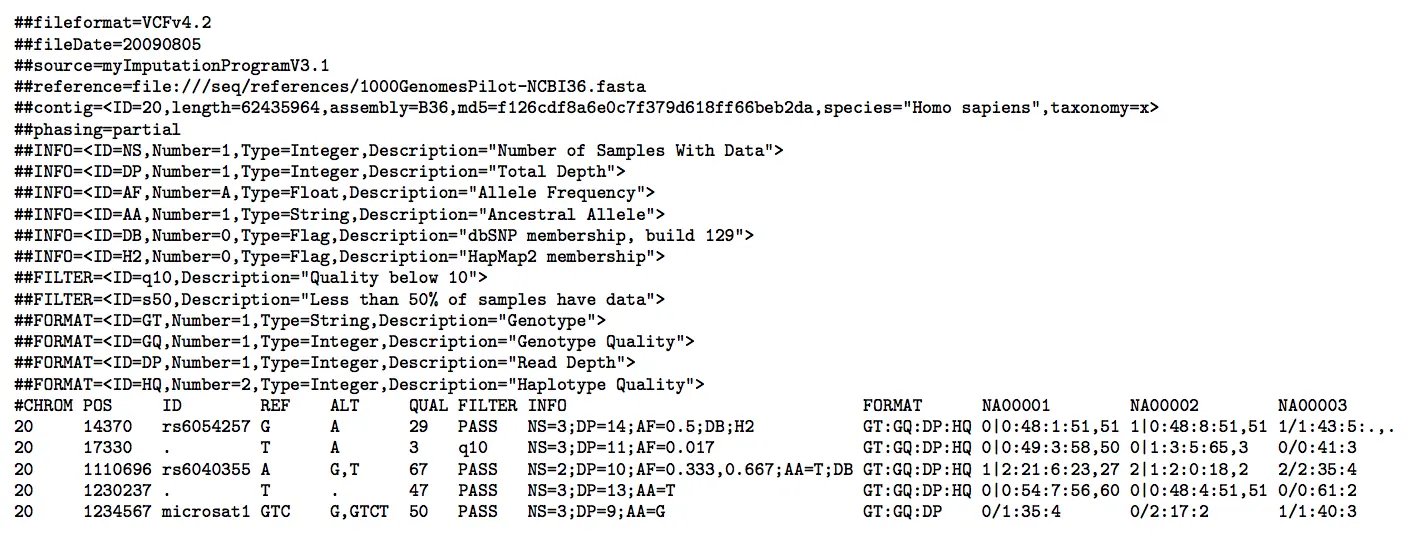
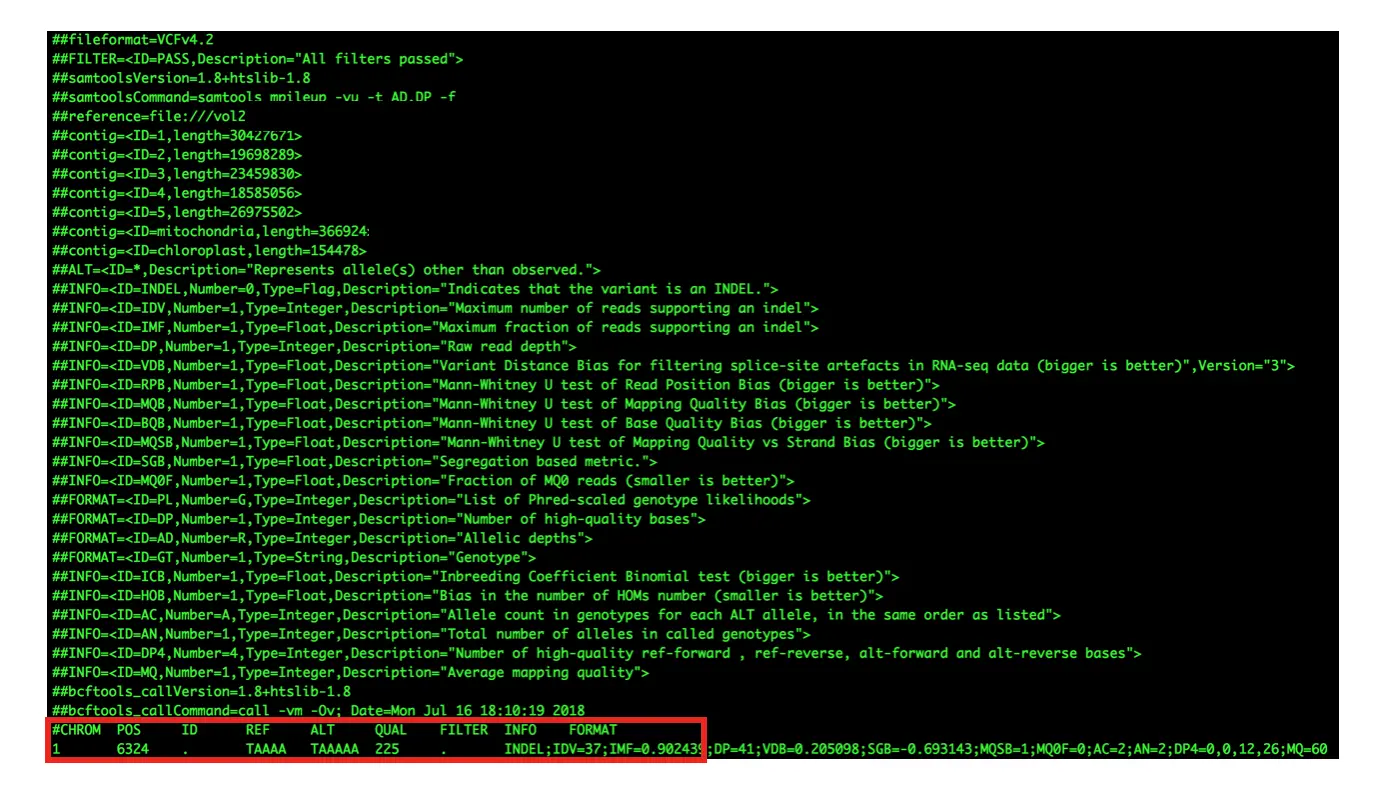
注释信息(##表示)
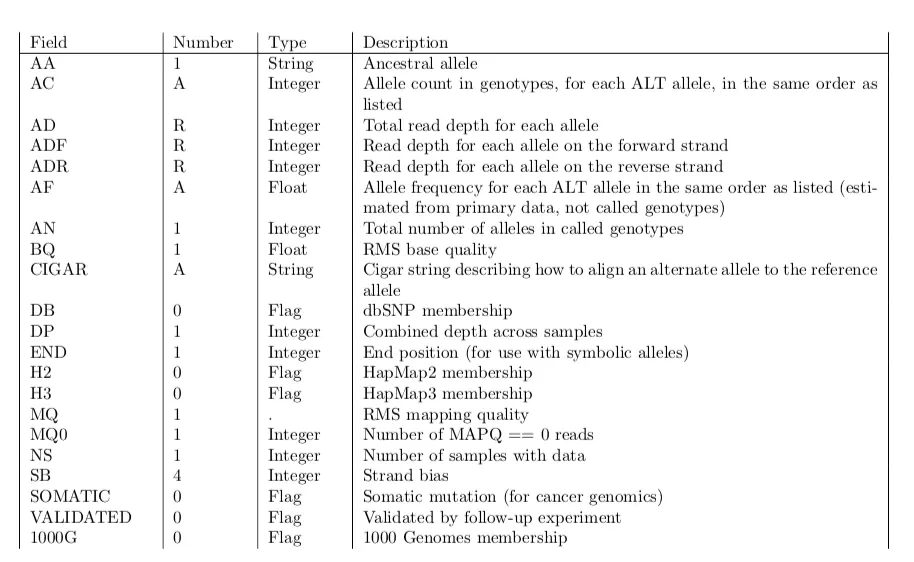
十分重要!后面每一个位点的描述的tag都在这个注释信息里面可以找到
INFO是碱基位点的注释,对应红框中的INFO,是对基因组特定位置进行的计算;FORMAT是每个样本都有的描述
- 第一行:一定是##fileformat ,VCF格式版本号
- ##FILTER:显示这个文件已经进行了过滤
- 然后是使用软件的名称及参数
- ##reference & contig:使用的参考基因组信息及参考基因组contig信息
- ##INFO行:每一行必须的四个标签是:ID、Number、Type、Description 主要有几个tag标记:AD、DP、GQ、GT、PL
具体信息
红框位置就是数据文件的头信息(#表示),主要有CHROM、POS、ID、REF、ALT、QUAL、FILTER、INFO、FORAMT、SAMPLE【前8列必须要有】
CHROM:变异位点从参考序列哪个染色体区段上找出来的
POS:异位点相对于参考基因组所在的最左端位置 (属于1-坐标系统:从1开始计数)【如果是InDel的情况,那么这个数值对应InDel的第一个碱基位置】
1-based coordinate system :序列的第一个碱基设为数字1,如SAM,VCF,GFF,wiggle格式 0-based coordinate system :序列的第一个碱基设为数字0,如BAM, BCFv2, BED, PSL格式
ID:变异位点名称(对应dbSNP数据库中的ID;若没有,则默认用
.表示他是一个novel variant)REF:参考序列该位置碱基类型及个数
ALT:该位置变异的碱基类型及个数【多个用逗号分隔;对于SNP是单个碱基的改变;对于InDel是碱基数量的改变】
QUAL:变异位点质量值(与测序数据一样也是用Phred格式表示)Phred值= -10 * log(1-P), P是变异位点存在的概率。值越大,此位点保持原状的概率越低,越可能发生变异。但是这个值随着数据量增大而变大,并非十分准确
FILTER:下一个位点是否要被过滤掉,如果显示PASS,说明下一个位点和参考序列一致,那么这个位点有更大可能性为变异位点
INFO:结合描述理解有关该位点的额外信息 【包含信息最多,形式为Tag=Value, 分号分隔】
FORMAT:变异位点格式
SMAPLE:使用的样本名称,由bam文件中@RG的SM标签决定
关于第八列INFO

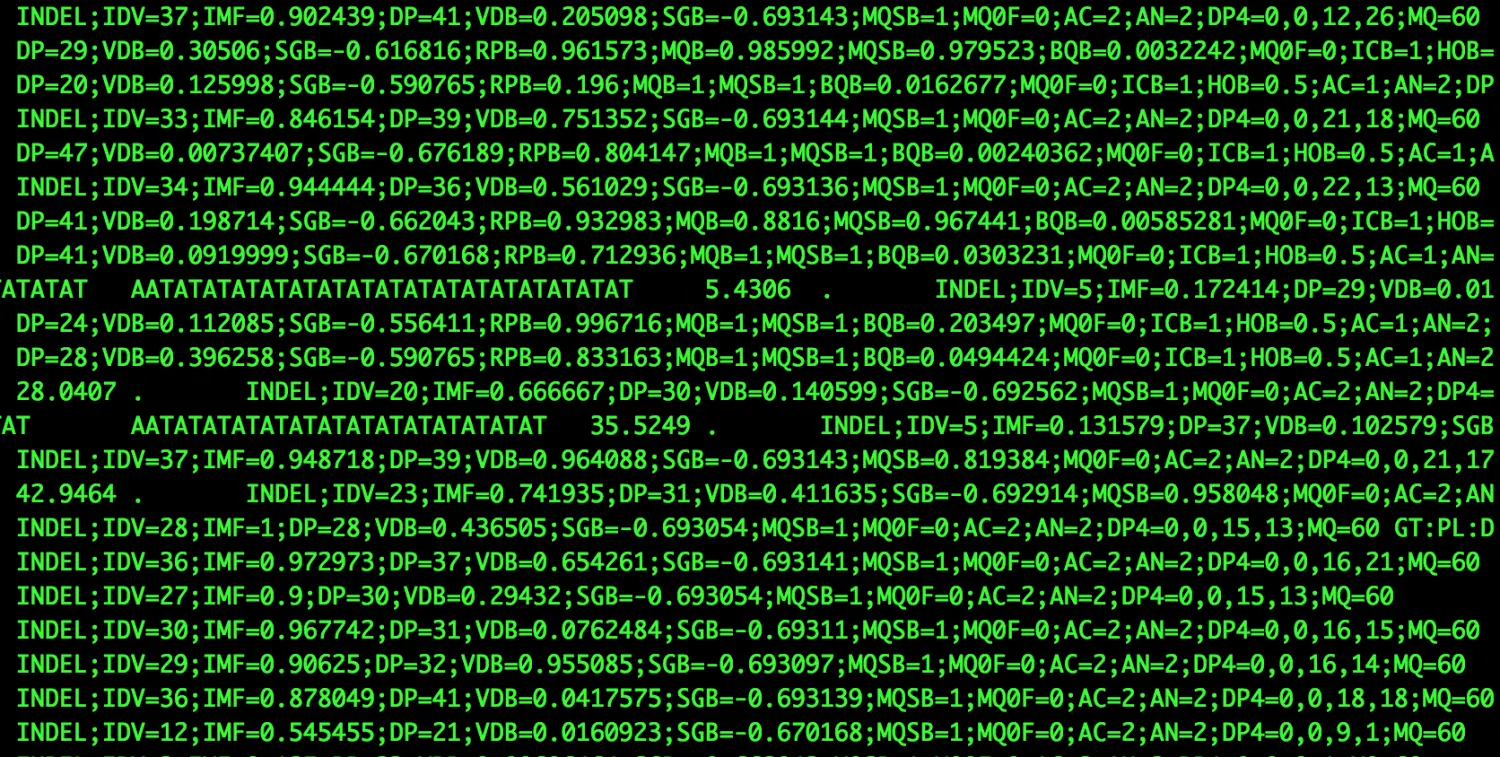
看上去是一列,但其中的内容可以无限扩增,常用的tag主要有:
AC、AF、AN【和等位基因有关】: AC:Allele Count该位点变异的等位基因数目; AF:Allel Frequency 等位基因频率; AN:Allel Number 等位基因的总数目
【单看这个不好理解,举一个二倍体diploid例子:基因型0/1表示为杂合子,该位点只有一个等位基因发生突变,AF=0.5(在该位点只有50%的等位基因发生突变),总的等位基因数目为2;基因型1/1表示为纯合子,AC=2,AF=1,AN=2】
DP:一部分reads被过滤掉后的覆盖度
DP4 : 高质量测序碱基,在ref或alt前后
Dels:官方解释是“Fraction of reads containing spanning delections”,这个值用来区分indel和snv【SNV与SNP?–一个物种中该单碱基变异的频率达到一定水平就叫SNP,而频率未知(比如仅仅在一个个体中发现)就叫SNV】 【有这个tag且为0时表示该位点是SNV,没有就是InDel,可以用来区分二者】
其他Tag:
还可以参考:http://www.bio-info-trainee.com/863.html
关于第九列FORMAT
表头的##FORMAT就是对第九列的解释,主要包括某一个特定位点基因型、测序深度的表述
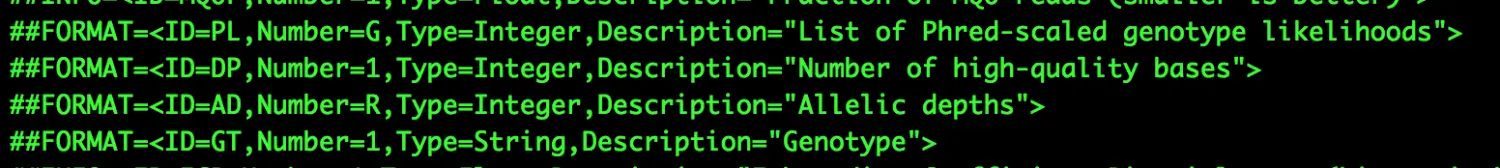
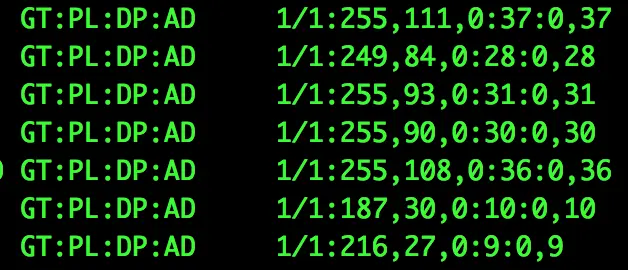
主要包含两列:前列为格式,后列为对应的数据【他们之间都用冒号隔开】
**GT:**样本基因型(genotype),两个数字之间【这里是1/1】斜线分隔,表示二倍体样本的基因型。0代表样本中ref的allel,1代表样本variant的allel,2表示有第二个variant的allel。0/0表示样本中该位点纯合,与ref一致;0/1表示样本中该位点杂合,有ref和variant两个基因型;1/1表示样本中位点纯合,与variant一致
AD和DP: 【第八列也有DP,但含义不同】 AD是Allele Depth,样本中每一种allel的reads覆盖度,在二倍体中是用逗号分隔的两个数,前面对应ref,后面对应variant; DP是Depth,是样本中该位点覆盖度
PL: Provides the likelihoods of the given genotypes指定三种基因型的质量值大小(基因型为0/0, 0/1, 1/1),对应的值越大,表示这种基因型的可能性越小