151-和刘小泽一起跟着官网学bedtools
刘小泽写于19.12.6 最近在学习ChIPseq分析,这个工具对数据格式转换和操作基因组坐标是很有帮助的。不过虽然很受欢迎,但还是有很多类似的工具可以用,比如 bedops。慢慢来,不着急,先学下这个目前引用超过8500的”瑞士军刀“
0 前言
这个工具的官网在:https://bedtools.readthedocs.io/en/latest/
作者 Aaron R. Quinlan 2010年把这个工具发在了Bioinformatics上,他当时还在美国弗吉尼亚大学医学院,现在工作在犹他大学【他的实验室链接:http://quinlanlab.org/,这个网站做的真心不错】
点开官网链接,它的logo早已说明一切。它支持对BAM、BED、GFF/GTF、VCF等和基因组区域有关文件的intersect, merge, count, complement, and shuffle等操作。或许这些名词还看不懂,没关系,一点点跟着例子走:

0 下面👇正文开始
官方教程在:http://quinlanlab.org/tutorials/bedtools/bedtools.html 我也是跟着这个学一遍,记录自己的心得
软件安装不多说,conda直接搞定
1 测试数据的下载
# 创建一个属于你自己的学习目录
mkdir -p ~/biosoft/bedtools-learn
cd ~/biosoft/bedtools-learn
# 然后下载测试数据,一个压缩包,一个txt,4个bed文件
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/maurano.dnaseI.tgz
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/cpg.bed
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/exons.bed
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/gwas.bed
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/genome.txt
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/hesc.chromHmm.bed
# 【当然,如果自己的网络不给力,也可以在公众号回复”bedtools“即可】
tar -zxvf maurano.dnaseI.tgz
rm maurano.dnaseI.tgz
0.1 这些数据都是什么?
- 可以看到解压后有20个以
f开头的bed文件(f 代表 fetal tissue 胎儿组织),表示来自大脑、心脏、肠、肾、肺、肌肉、皮肤和胃的20个不同胎儿组织样本。 cpg.bed表示人类基因组的CpG岛exons.bed表示来自RefSeq的人类基因外显子.gwas.bed表示用GWAS鉴定的与人类疾病相关的SNPshesc.chromHmm.bed表示基于ENCODE的ChIP-seq实验,利用chromHMM预测的人类胚胎干细胞基因组区间的功能
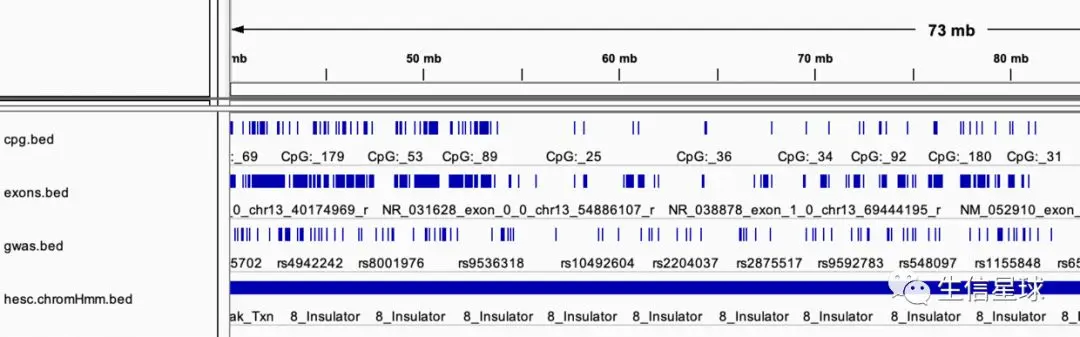
这四个文件都是从:UCSC Table Browser 获得的,把它们导入IGV是这样:
0.2 bedtools概况
这个工具有几十个子命令,比如使用时要输入:
bedtools intersect
bedtools merge
bedtools subtract
...
查看版本:
bedtools --version
强烈建议下面的命令自己手动敲一遍!
1 第一个最常用的:bedtools intersect
它的作用是比较两个或多个BED/BAM/VCF/GFF,然后找它们重叠的区域(就是指至少存在1个公共的碱基)
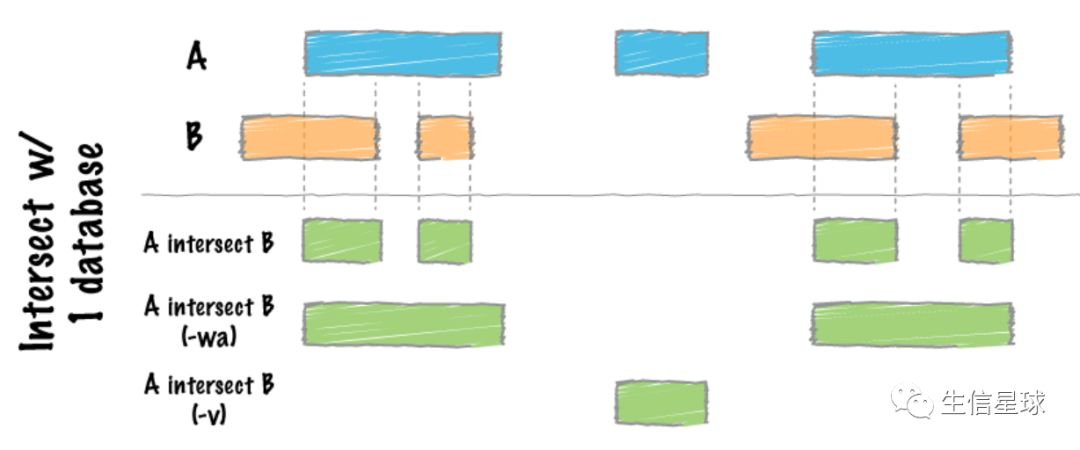
1.1 官方给出的手绘图感觉最好理解
如果只比较两个文件 A和B
解释一下”绿色部分的结果“
- 第一种没有参数的:最好理解,就是简单的返回A和B共有的区域
- 第二种加了
-wa参数:它的含义是Write the original entry in A for each overlap,也就是把原始A文件的坐标输出来 - 第三种加了
-v参数:只输出A中有且B中没有的区域(很像grep -v)
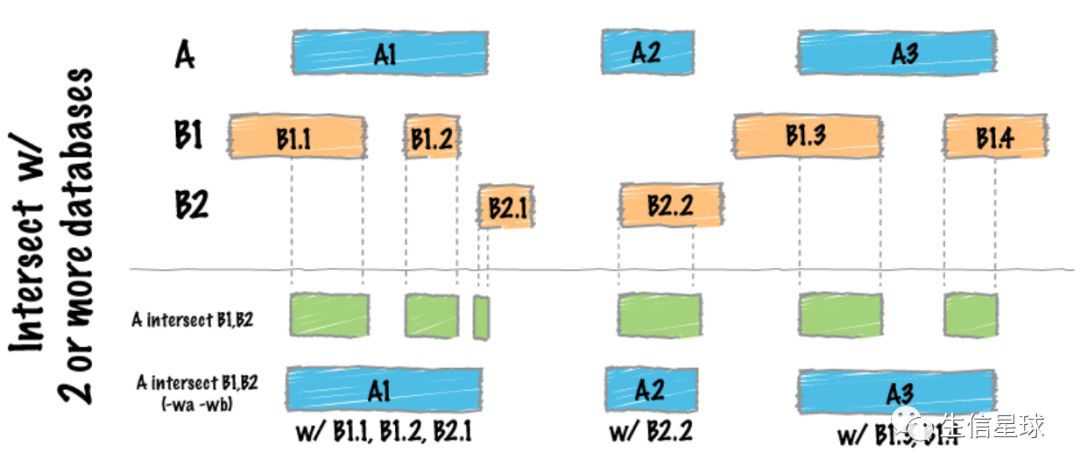
如果比较多个文件 A 和 B1、B2
- 第一种结果:输出A与B1、B2三者共有的区域
- 第二种加了
-wa、-wb参数:先输出A的坐标,然后输出与A有重叠的B1、B2的区域坐标
1.2 默认的输出结果
通过-a、-b指定文件A、B
bedtools intersect -a cpg.bed -b exons.bed | head -5
chr1 29320 29370 CpG:_116
chr1 135124 135563 CpG:_30
chr1 327790 328229 CpG:_29
chr1 327790 328229 CpG:_29
chr1 327790 328229 CpG:_29
需要注意的是:这里打印出来的结果并不是原始的CpG记录的区间,而是它和exon.bed的重叠区间,因此坐标只是原来CpG.bed的子集
1.3 想看具体重叠双方的区域
Not only showing you where the intersections occurred, it shows you what intersected
bedtools intersect -a cpg.bed -b exons.bed -wa -wb | head -5
chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r 0 -
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 -
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 +
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 +
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 +
就第一行结果来说:
# 3.2中得到的是
chr1 29320 29370 CpG:_116
# 3.3 得到的是
chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r 0 -
看到原始的CpG记录是28735 - 29810,只不过有一个外显子坐标(29320 - 29370)包含其中,于是3.2的结果只记录了29320 - 29370
1.4 想统计重叠的碱基数
使用参数-wo (write overlap),会在每一行结果末尾统计这个重叠区域有多长
例如:下面👇结果结尾的50、439
bedtools intersect -a cpg.bed -b exons.bed -wo | head -5
chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r 0 - 50
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 - 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 + 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 + 439
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 + 439
1.5 统计发生重叠的features数量
这个feature的含义很广,不限于基因,还可以是外显子、转录本等等与基因组有关的信息
使用参数-c ,就会统计对文件A中的每个feature,在文件B中有多少个features与之有交集
例如:下面👇结果结尾的1、1、3、0、0
bedtools intersect -a cpg.bed -b exons.bed -c | head -5
chr1 28735 29810 CpG:_116 1
chr1 135124 135563 CpG:_30 1
chr1 327790 328229 CpG:_29 3
chr1 437151 438164 CpG:_84 0
chr1 449273 450544 CpG:_99 0
1.6 统计没有重叠的区域
使用-v参数【这个应该很容易理解】
bedtools intersect -a cpg.bed -b exons.bed -v | head -5
chr1 437151 438164 CpG:_84
chr1 449273 450544 CpG:_99
chr1 533219 534114 CpG:_94
chr1 544738 546649 CpG:_171
chr1 801975 802338 CpG:_24
1.7 设定一个最小重叠比例
之前说过,软件默认只要二者有1bp交叉,也算重叠区域。但这个我们可以自行调整。
例如,设定至少满足文件A的百分之50要与文件B有重叠,才统计。使用参数-f (fraction)
bedtools intersect -a cpg.bed -b exons.bed -wo -f 0.5 | head -5
chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 - 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 + 439
chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 + 439
chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 + 439
chr1 788863 789211 CpG:_28 chr1 788770 794826 NR_047519_exon_5_0_chr1_788771_f 0 + 348
然后以第一行结果为例,在IGV中查看,发现这个确实是满足的,因为文件A(cpg.bed)的100%都落在了文件B(exons.bed)中:
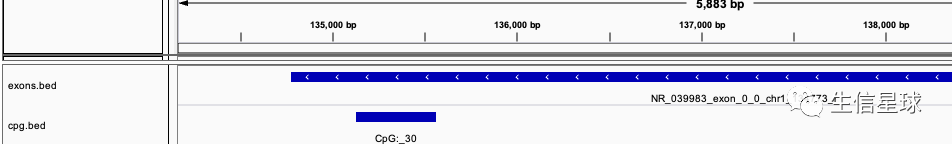
1.8 「小贴士」sort后的数据运行的更快哦😯
sort的含义是:对染色体和起始坐标进行排序(比如最简单的命令:sort -k1,1 -k2,2n;当然对bam文件排序可以用samtools的sort命令)
比如作者在这里做了一个演示:
time bedtools intersect -a gwas.bed -b hesc.chromHmm.bed > /dev/null
1.10s user 0.11s system 99% cpu 1.206 total
time bedtools intersect -a gwas.bed -b hesc.chromHmm.bed -sorted > /dev/null
0.36s user 0.01s system 99% cpu 0.368 total
而且文件越大,sort的优势越明显,也许这正是得到bam文件后sort的用意吧
1.9 一次寻找多个文件的重叠区域
在bedtools的2.21.0版本之后,便可以实现参数-b指定多个文件,大大简化了一次实验多个数据之间的比较
例如:寻找外显子与 CpG islands、GWAS SNPs、ChromHMM annotations的交叉【记得进行sort哦😯】:
bedtools intersect -a exons.bed -b cpg.bed gwas.bed hesc.chromHmm.bed -sorted | head -5
chr1 11873 11937 NR_046018_exon_0_0_chr1_11874_f 0 +
chr1 11937 12137 NR_046018_exon_0_0_chr1_11874_f 0 +
chr1 12137 12227 NR_046018_exon_0_0_chr1_11874_f 0 +
chr1 12612 12721 NR_046018_exon_1_0_chr1_12613_f 0 +
chr1 13220 14137 NR_046018_exon_2_0_chr1_13221_f 0 +
不过,这样我们看不到 文件(B1、B2…)与文件A到底在哪里产生了重叠
方案一:加上-wa、-wb参数
bedtools intersect -a exons.bed -b cpg.bed gwas.bed hesc.chromHmm.bed -sorted -wa -wb \
| head -10000 \
| tail -5
看到结果的第7列记录了一些数字:这代表了是文件B中的第几个文件(比如这个3 就表示hesc.chromHmm.bed这个文件,说明它和文件A exons.bed 存在交集);另外,如果多个文件B与文件A的相同区域有交集,那么也是每一行记录一个文件

方案二:可能用数字表示文件还是不直观,我们还要回过头去查看到底是哪个文件
于是可以用参数-names提前给多个文件B起绰号
bedtools intersect -a exons.bed -b cpg.bed gwas.bed hesc.chromHmm.bed -sorted -wa -wb -names cpg gwas chromhmm \
| head -10000 \
| tail -10
再看一下结果,这样是不是就方便查看了?
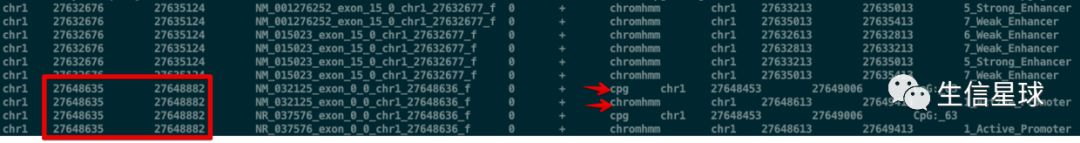
方案三:更直接的用文件名表示
bedtools intersect -a exons.bed -b cpg.bed gwas.bed hesc.chromHmm.bed -sorted -wa -wb -filenames -sorted
# 结果会在中间显示我们的query与target中的哪个文件存在交集
2 第二个常用的:bedtools merge
许多数据集的基因组feature坐标经常是连续的(比如ChIPseq的结果),就像下图的蓝色部分
于是可以把这些连续的基因组小区间连接起来,拼成一个连续的大区间
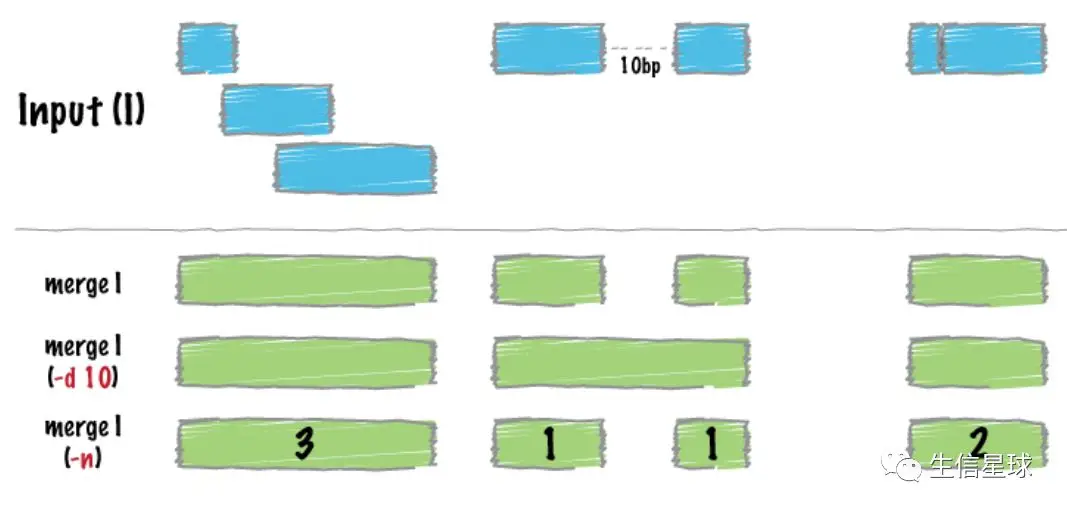
需要拼接的输入文件(bed/gff/vcf)必须是排序(sort)过的【不sort会报错🙅♂️】
先按染色体,再按起始位点,这样保证merge的算法执行起来非常顺畅,而基本不需要消耗内存再次加工
# 这样操作
sort -k1,1 -k2,2n foo.bed > foo.sort.bed
2.1 以exon.bed为例,展示merge的作用
head -n10 exons.bed
注意看:第3行和第4行的区间是有重叠的,因此它们可以进行merge
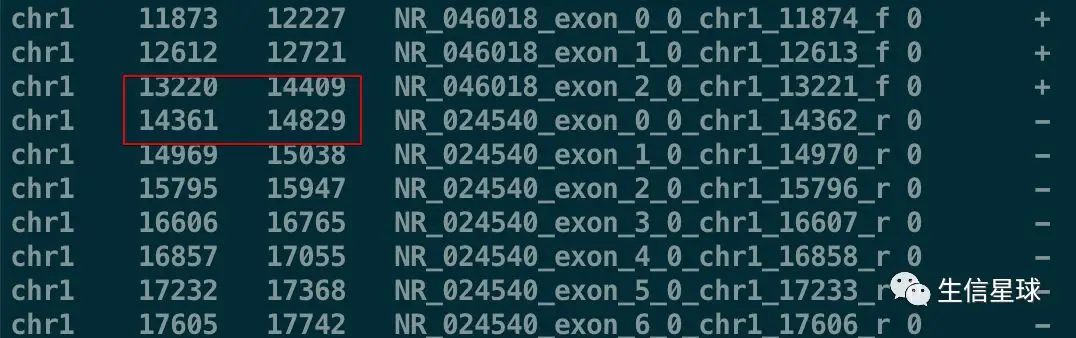
bedtools merge -i exons.bed | head -10
可以看到,merge之后原来的第3行(13220 - 14409)和第4行(14361 - 14829)坐标合并成了13220 - 14829
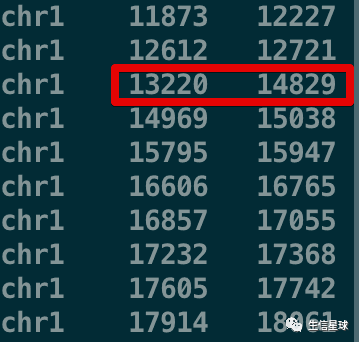
2.2 merge后统计之前重叠区间的数量
就像上面👆的第3行和第4行,它们合并在一起后,我们希望能直观看出新的区间13220 - 14829 是不是合并后的;如果是,合并了多少重叠的区域?
使用参数:-c和-o,其中-c指定按照第几列进行统计;-o指定计算方法(默认是sum)
例如:统计数量可以指定-o为count,此时按照第几列统计都是一样的【但如果指定-o sum,那么指定的列必须是数值,例如第2列 ”start position“,结果就会把重叠区域的start position 相加】
# 仅仅统计次数就用count【可以指定第一列chr,但实际上还是按第2-3列的坐标进行识别】
bedtools merge -i exons.bed -c 1 -o count | head -n 10
chr1 11873 12227 1
chr1 12612 12721 1
chr1 13220 14829 2
chr1 14969 15038 1
chr1 15795 15947 1
chr1 16606 16765 1
chr1 16857 17055 1
chr1 17232 17368 1
chr1 17605 17742 1
chr1 17914 18061 1
# 默认是sum【此时就要指定第2列或其他数值型的列】
bedtools merge -i exons.bed -c 2 -o sum | head -n 10
chr1 11873 12227 11873
chr1 12612 12721 12612
chr1 13220 14829 27581
chr1 14969 15038 14969
chr1 15795 15947 15795
chr1 16606 16765 16606
chr1 16857 17055 16857
chr1 17232 17368 17232
chr1 17605 17742 17605
chr1 17914 18061 17914
2.3 即使不重叠,也能merge
使用-d参数(distance),就像第4部分刚开始一样,指定了 -d 10,一开始相距10bp的两个区间也合并在了一起。
再举个例子:如果认为相聚不超过1000bp的两个区间,依然表示同一个,那么也可以合并
bedtools merge -i exons.bed -d 1000 -c 1 -o count | head -10
chr1 11873 18366 12
chr1 24737 24891 1
chr1 29320 29370 1
chr1 34610 36081 6
chr1 69090 70008 1
chr1 134772 140566 3
chr1 323891 328581 10
chr1 367658 368597 3
chr1 621095 622034 3
chr1 661138 665731 3
2.4 merge了这么多,我怎么知道是哪些?
还是借助上面提到的-o参数,它除了能统计个数、计算值的大小,还能输出名称【使用collapse】。只要给它指定了对哪一列进行操作
例如:exon.bed中的第4列是exon的名称,这样在count结束后,就可以再根据第4列输出名称【如果存在合并的现象,那么其中所有的exon都会列出来,并以逗号分隔】
bedtools merge -i exons.bed -d 90 -c 1,4 -o count,collapse | head -10
chr1 11873 12227 1 NR_046018_exon_0_0_chr1_11874_f
chr1 12612 12721 1 NR_046018_exon_1_0_chr1_12613_f
chr1 13220 14829 2 NR_046018_exon_2_0_chr1_13221_f,NR_024540_exon_0_0_chr1_14362_r
chr1 14969 15038 1 NR_024540_exon_1_0_chr1_14970_r
chr1 15795 15947 1 NR_024540_exon_2_0_chr1_15796_r
chr1 16606 16765 1 NR_024540_exon_3_0_chr1_16607_r
chr1 16857 17055 1 NR_024540_exon_4_0_chr1_16858_r
chr1 17232 17368 1 NR_024540_exon_5_0_chr1_17233_r
chr1 17605 17742 1 NR_024540_exon_6_0_chr1_17606_r
chr1 17914 18061 1 NR_024540_exon_7_0_chr1_17915_r
3 学习第三个:bedtools complement 实现反选
给定一个feature坐标信息文件,我们如果不关心其中标记的区间,而是想看看有哪些区间不在这个文件中
例如,有一个ChIP-seq的peaks信息,现在想知道有哪些区域没有被抗体结合,就可以用complement
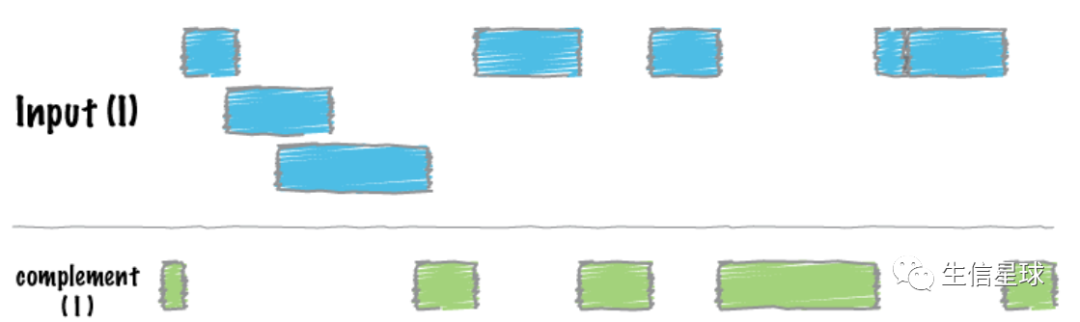
现在有了外显子的bed文件,通过反选,我们就能获得内含子或基因间区的坐标。那么既然是反选,除了exon.bed,还要有一个总体的范围(也就是基因组各个染色体的长度信息)【用-g指定】
# 首先要确定整体的操作范围
head -10 genome.txt
chr1 249250621
chr10 135534747
chr11 135006516
chr11_gl000202_random 40103
chr12 133851895
chr13 115169878
chr14 107349540
chr15 102531392
chr16 90354753
chr17 81195210
# 实现反选,选出不属于exon的区域
bedtools complement -i exons.bed -g genome.txt | head -10
chr1 0 11873
chr1 12227 12612
chr1 12721 13220
chr1 14829 14969
chr1 15038 15795
chr1 15947 16606
chr1 16765 16857
chr1 17055 17232
chr1 17368 17605
chr1 17742 17914
看到Chr1的第一个外显子就是从11874开始
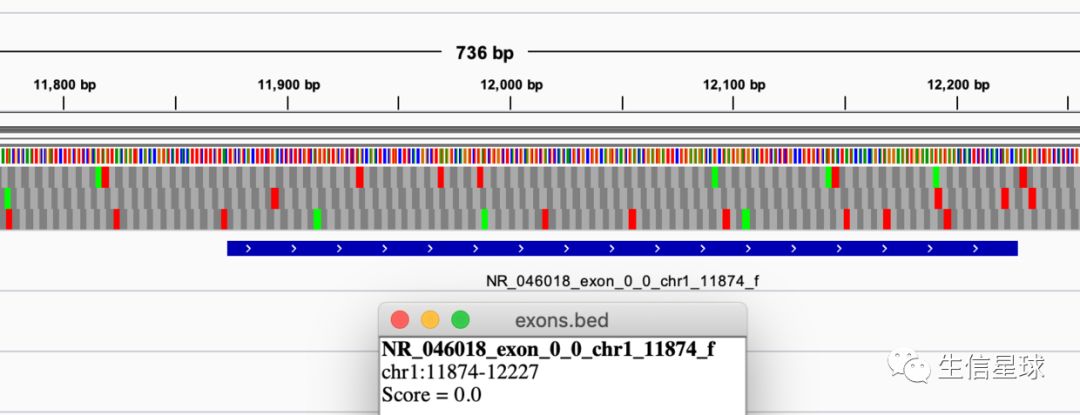
但是不知道你注意没有,前面exon.bed中记录的chr1的第一个外显子坐标是
chr1 11873 12227 1 NR_046018_exon_0_0_chr1_11874_f并非是11874开始,这是因为:
坐标系统 :
- 1-based coordinate system :序列的第一个碱基设为数字1,如SAM,VCF,GFF,wiggle格式
- 0-based coordinate system :序列的第一个碱基设为数字0,如BAM, BCFv2, BED, PSL格式
6 学习第四个:bedtools genomecov 计算基因组覆盖度和深度
这个功能看上去摸不着头脑,实际上不难并且很实用
最简单的命令是:
bedtools genomecov -i exons.bed -g genome.txt
这个命令的默认输入是bed/gff/vcf ,并且需要加一个参考基因组的染色体大小记录文件。
6.1 来看一张关于genomecov 的图
看到这张图,第一反应是不是:这些0、1、2、3哪来的,有什么用?【没关系,一起探索】
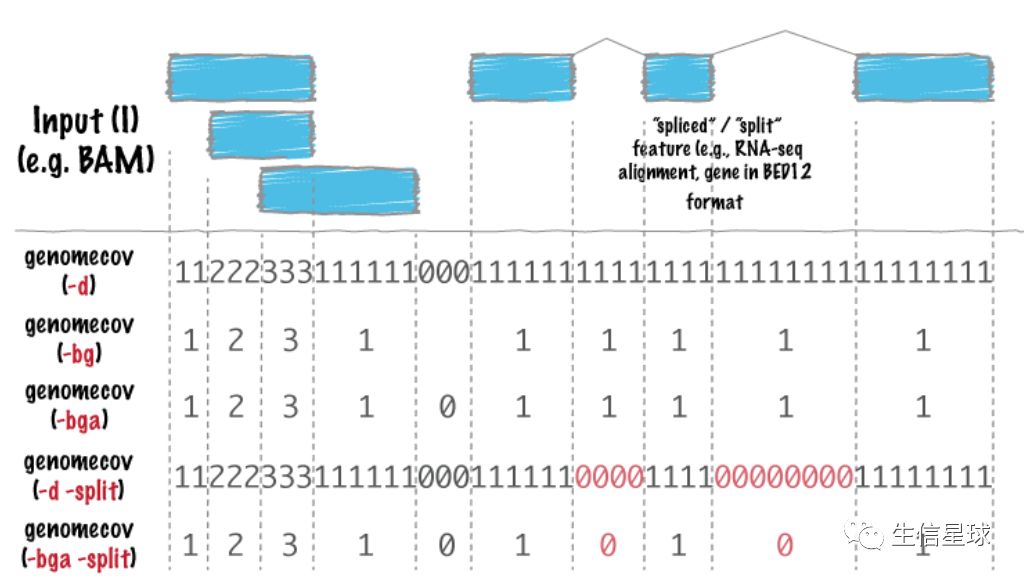
6.2 什么是测序深度?
我们测序(尤其是ChIPseq),比较重要的一个目的就是查看测序深度,比如这些:
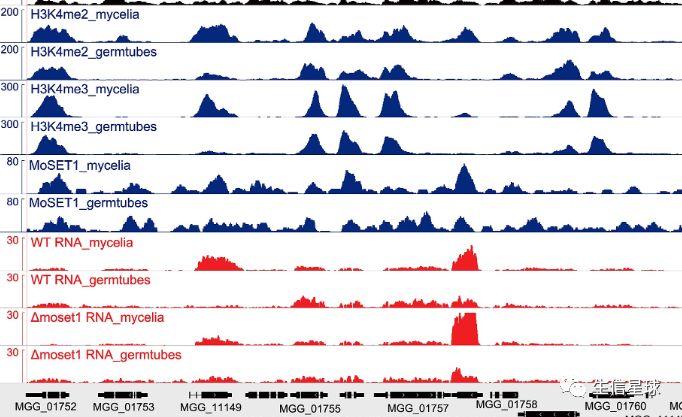
大家肯定对sam、bam文件不陌生,其中就记录了测序reads比对到基因组上的位置信息
【想对sam、bam文件补课?可以看: 处理SAM、BAM你需要Samtools】
那么根据这个信息,就能反推基因组上每个碱基所对应的reads数,当然不用我们手动计算,samtools帮我们一把。可以看到下面得到的这个测序深度文件test.depth,第一列是染色体编号,第二列是染色体上碱基的位置,第三列是覆盖到这个位点的reads数目:
# 以果蝇的bam数据为例
samtools depth dm.sort.bam > test.depth
head -5 test.depth
2L 40 1
2L 41 1
2L 42 1
2L 43 1
2L 44 1
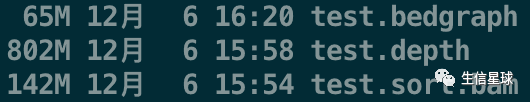
不过,虽然这个文件很易读,但每个碱基都记录于此,文件岂不是太大?(以人类为例,就需要记录30亿个碱基的信息,十分的冗余,因此得到的depth文件一般要比bam文件还要大几倍)
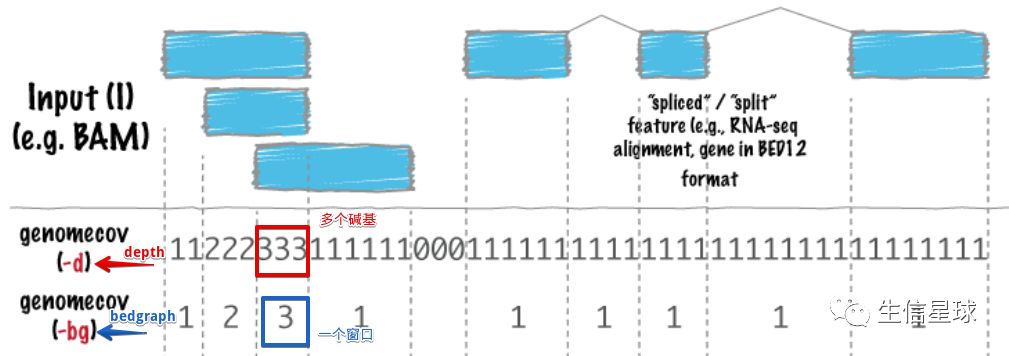
于是,为了体现测序深度的同时节省空间,又开发了两种新的文件格式:bedGraph和wiggle
6.3 bedGraph是什么?和depth文件有什么区别?
首先来看bedGraph文件的产生:
bedtools genomecov -ibam dm.sort.bam -bg > test.bedgraph
如果要指定一个bam作为输入,就不需要使用-i和-g参数了。可以直接用-ibam指定输入bam文件;
然后-bg表示产生bedgraph文件
如果使用
-bga就表示:将feature之间的间隔区域记做窗口0(而-bg的结果中,这部分是不记录的)
看下它的内容
head test.bedgraph
2L 39 65 1
2L 65 66 2
2L 66 90 3
2L 90 116 2
2L 116 117 1
2L 255 256 1
2L 256 261 2
2L 261 284 3
2L 284 306 4
2L 306 307 3
区别
bedgraph文件将之前depth文件的每个碱基测序深度整合成为一个个的窗口,第四列记录的就是窗口中的测序深度,再看那个图,可以看到bedgraph将原来depth中具有相同测序深度的连续的碱基,合并成了一个小窗口:
最后,看一眼文件的大小差异
7 如果你认为bedtools只能做这些,那就不是瑞士军刀了
它的小功能应该有几十个,但最应该学习的是处理思想 官方给的建议是:Analytical power in
bedtoolscomes from the ability to “chain” together multiple tools in order to construct rather sophisicated analyses with very little programming - you just need genome arithmetic!
下面将介绍不同的例子,来增加对它的认识
例1
给定一个a.bam和b.regions.bed,现在想看看region中的坐标在bam中没有体现的那部分
思路很清晰:先找bam中没有覆盖到的区域(用-bga 标记这部分为0),然后把0挑出来就是不在bam上的区域;然后intersect解决
bedtools genomecov -ibam a.bam -bga | awk '$4==0' | bedtools intersect -a regions -b - >result
例2
检查bam文件中比对到外显子的覆盖率
bedtools bamtobed -i reads.bam | \
bedtools coverage -a - -b exons.bed \
> exons.bed.coverage
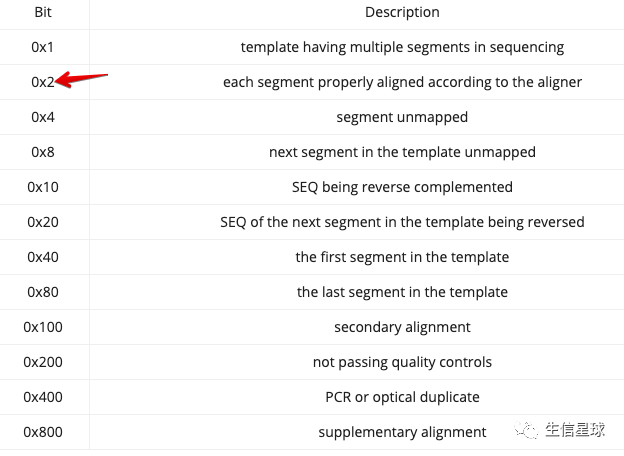
再进一步, 查看properly-paired reads的覆盖度
这里的-uf 0x2 目的就是提取出来bam中的properly-paired reads,只不过用来二进制表示
关于flags可以看:https://davetang.org/muse/2014/03/06/understanding-bam-flags/
samtools view -uf 0x2 reads.bam | \
coverageBed -abam - -b exons.bed \
> exons.bed.proper.coverage
例3
对每条链分别计算覆盖度
# 先看+链(重点是根据转换的bed文件中的+/-那一列进行区分)
bedtools bamtobed -i reads.bam | \
grep \+$ | \
bedtools coverage -a - -b genes.bed \
> genes.bed.forward.coverage
# 然后看-链
bedtools bamtobed -i reads.bam | \
grep \-$ | \
bedtools coverage -a - -b genes.bed \
> genes.bed.reverse.coverage
例4
对自己记录的SNPs位点进行筛选,找到那些既不在dbSNP也不在1000 genomes中的
思路:核心就是对bed进行过滤(-v参数)
bedtools intersect -a snp.calls.bed -b dbSnp.bed -v | \
bedtools intersect -a - -b 1KG.bed -v | \
> snp.calls.novel.bed
例5
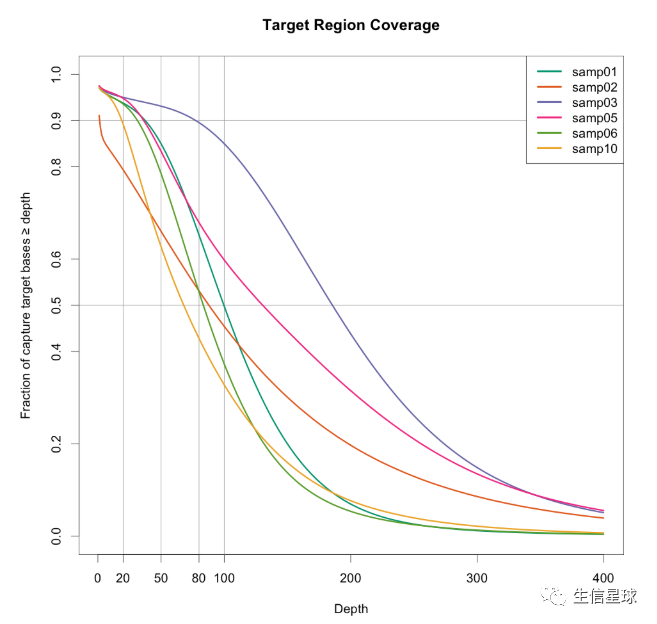
对外显子捕获效率的可视化
这个检测的标准可以参考:For example, covering 90% of the target region at 20X coverage may be one metric to assess your ability to reliably detect heterozygotes
思路:有一个测序reads比对的bam文件,有一个探针捕获区域的bed文件(比如:
NimbleGen’s V3 exome capture regions here ),然后用bedtools coverage命令,加-hist参数表示输出bed文件中各个feature在bam上覆盖度的直方图结果
直接看代码:https://www.gettinggeneticsdone.com/2014/03/visualize-coverage-exome-targeted-ngs-bedtools.html
例8
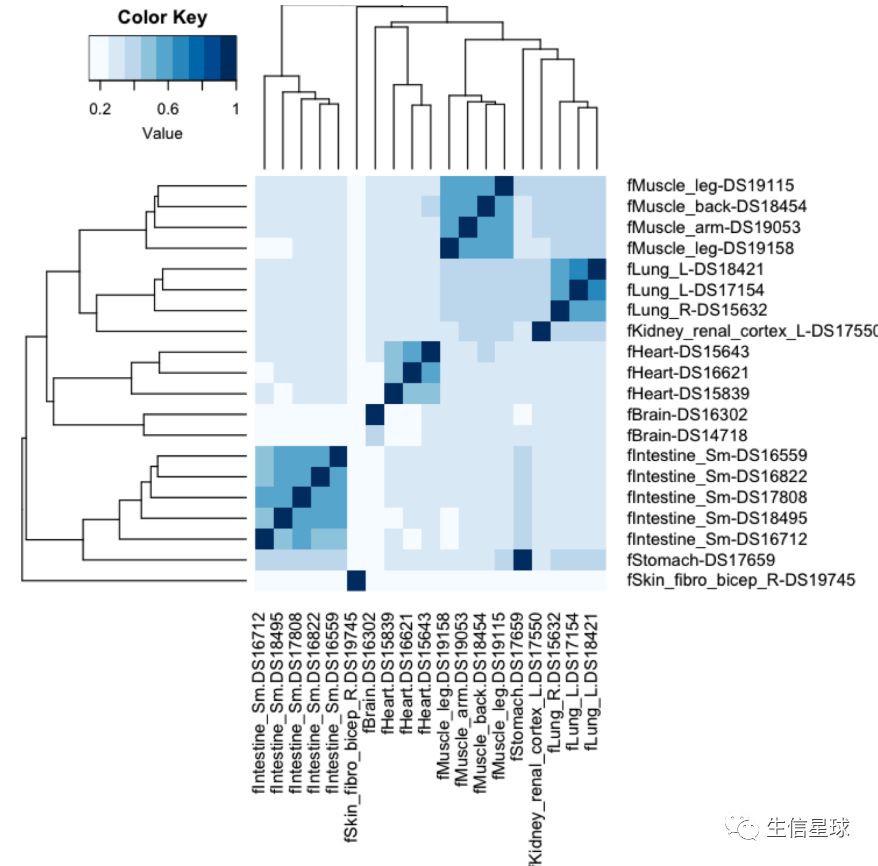
检测两个数据之间的相关性
引入一个新的bedtools工具jaccard,它会计算一个杰卡德相似性系数
结果是0.0 to 1. 0的值,数越小相关性越小
# 检测同一个样本的不同数据【系数是0.50637】
bedtools jaccard \
-a fHeart-DS16621.hotspot.twopass.fdr0.05.merge.bed \
-b fHeart-DS15839.hotspot.twopass.fdr0.05.merge.bed
intersection union-intersection jaccard n_intersections
81269248 160493950 0.50637 130852
# 再看不同的样本的不同数据【系数是0.170995】
bedtools jaccard \
-a fHeart-DS16621.hotspot.twopass.fdr0.05.merge.bed \
-b fSkin_fibro_bicep_R-DS19745.hg19.hotspot.twopass.fdr0.05.merge.bed
intersection union-intersection jaccard n_intersections
28076951 164197278 0.170995 73261
另外,还能分析更多的样本之间相关性
这个就看(官网http://quinlanlab.org/tutorials/bedtools/bedtools.html)翻到最底部
小结
叫小结一点都不夸张,上面的内容都是初级内容,是带领我们熟悉这个工具。就像搭积木,工具简单,但实现的方法千千万。
bedtools全部小功能列表
其中每一个小功能都有自己的链接,点进去,又是一片天地
https://bedtools.readthedocs.io/en/latest/content/bedtools-suite.html
bedtools的示范操作
帮你快速记忆,有点像cheatsheet
https://bedtools.readthedocs.io/en/latest/content/example-usage.html
常见的一些问题
很多都是安装相关的,既然用conda安装,这里就省去了很多麻烦