043-来一场Cytoscape的旅行
刘小泽写于18.10.13
这是我第一次使用这个工具,之前下载过但并不知道怎么用,只是出于对强大工具的好奇。但这次必须要用到了,前面做了WGCNA,得到了一些hub genes,那么就要对这些基因进行网络可视化。哪来的自信让自己去学习一个陌生的工具呢?是美感!我们都是视觉动物,一旦被它的强大所征服,什么英文文档,什么操作流程,都不再是我们的羁绊而是牵引力。并且一定要相信,工具做出来一定不难使用,否则作者都对不起他自己的劳动,只是偶尔我们需要和作者进行心灵的沟通。May the force be with you!
Cytoscape是什么
它诞生于2003年,旨在为用户提供开源的网络可视化的工具。现在研究单一因素已经不能说服别人了,需要多个实验、多个组学共同表明,比如研究基因共表达、miRNA-gene互作、蛋白互作、菌落互作、靶向调控等。引用量惊人1W+
得到它
下载地址:https://cytoscape.org/download.html,需要Java8(再高的版本就不支持了)没有的话也不用担心,安装程序会自动帮你下载。软件最大的特色就是支持多种插件(就像chrome为什么好用,就是因为插件齐全);缺点就是比较消耗内存。
初次见面
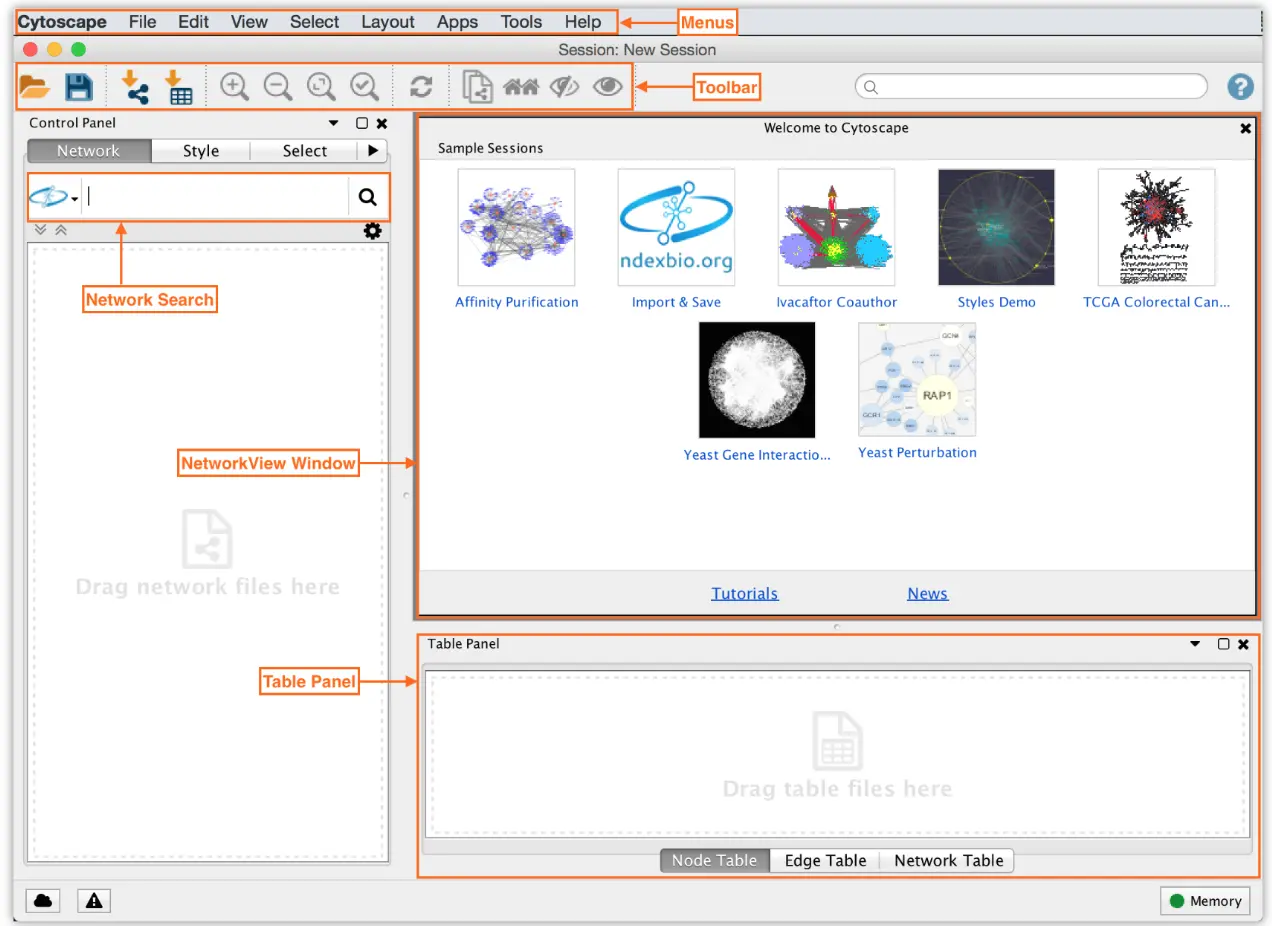
Menus:菜单栏(常用的几个介绍)
View:显示或者隐藏上图的几大板块
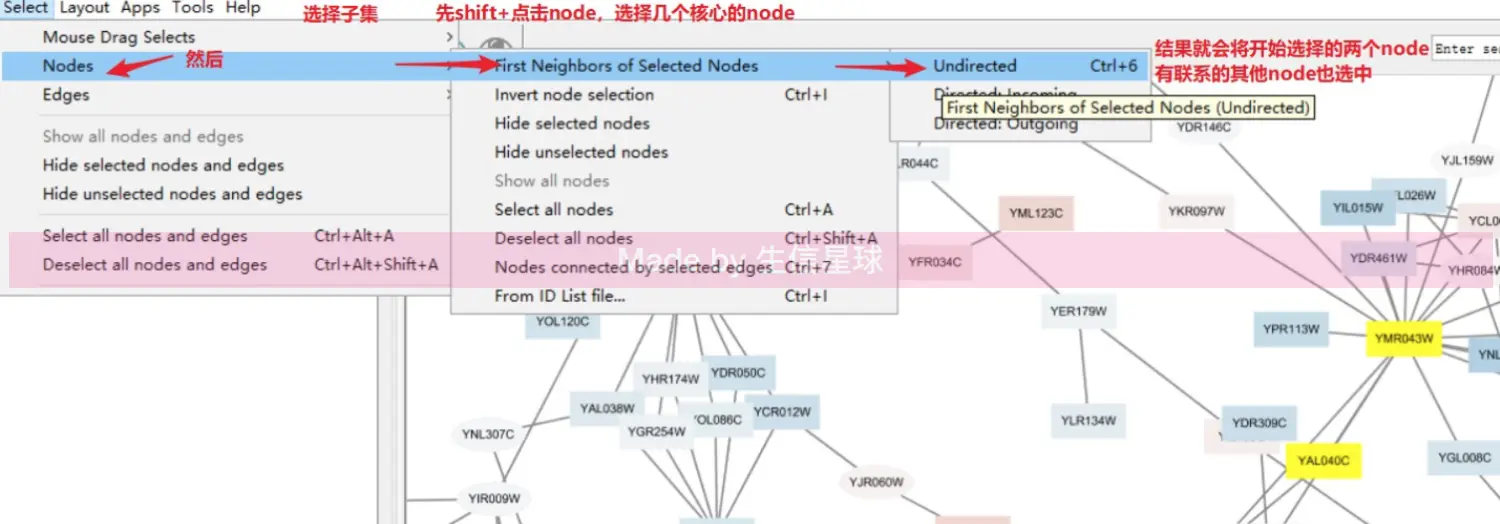
Select:支持快速选择相邻的所有节点、线
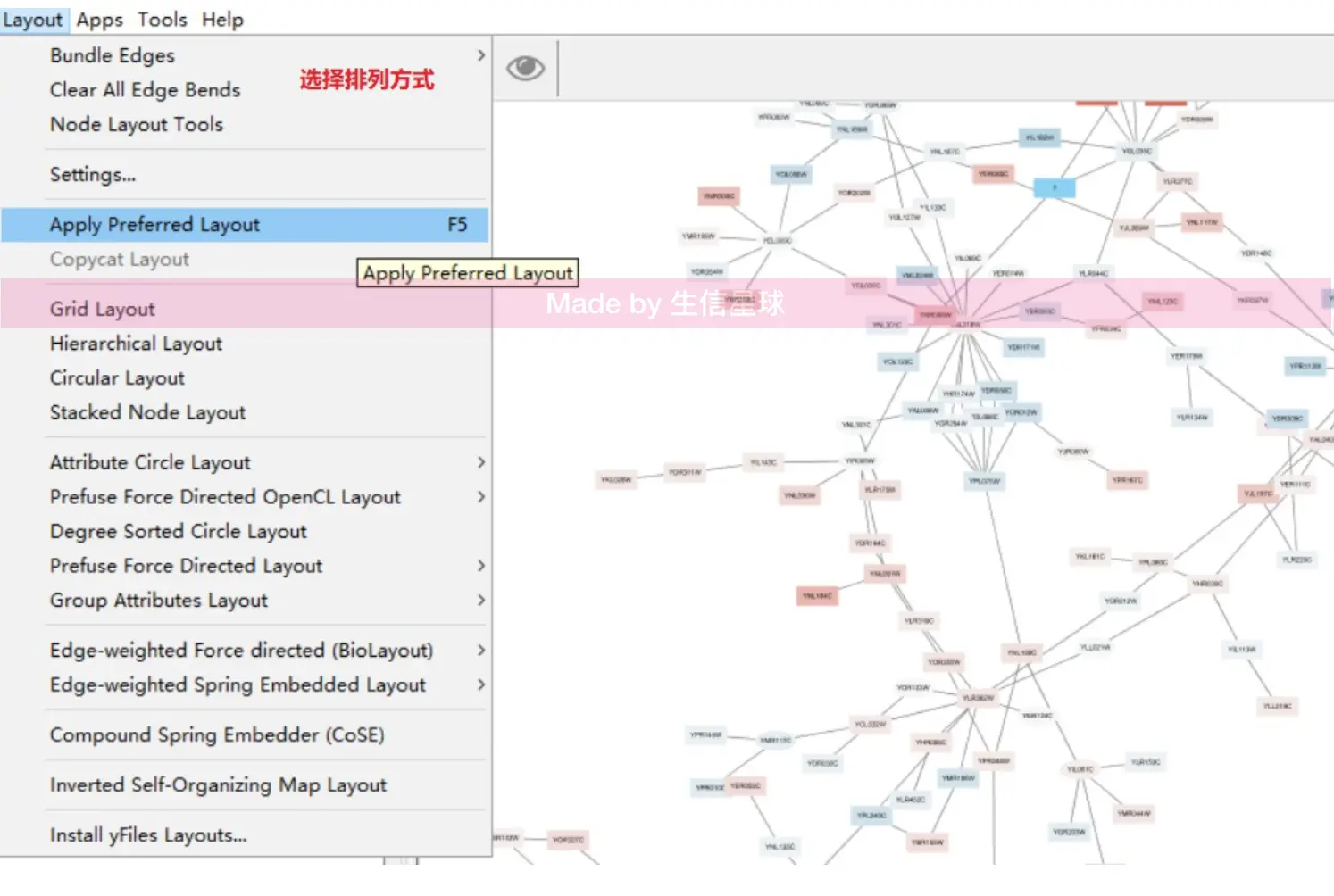
Layout:多种网络形态供选择;还可以在Node Layout tools中对网络进行旋转(rotate)、拉伸(scale);对线(edge)弯曲;恢复成原始面貌(Apply Preferred Layout)
scale选项可以避免太多的节点凑在一起,可以让它们分散一下 edge选项实现的线弯曲,有时可以让图更美观
App:这一个是软件的重点
Toolbar:工具栏(许多的快捷操作)

Network Search:主要的参数调整区域
- Network
- Style:设置包括三部分:node,edge,network
- Select
NetworkView Window:看图区
Table Panel:显示节点Node、线Edge、网络Network的属性
还有右下角的内存检测(红黄绿三种颜色,绿色表示内存充足)
敬个礼握握手
相关知识
- 节点node:相互作用的分子如基因、蛋白等;
- 线/边edeg:连接两个节点,表示其相互作用;
- 度degree:与某个节点相互作用的节点数量,度越大,节点的核心程度越高;因此如果一个基因分布的通路较多,那么它的度就大,显示的点就大
先来一个简单的流程,获得点成就感再说
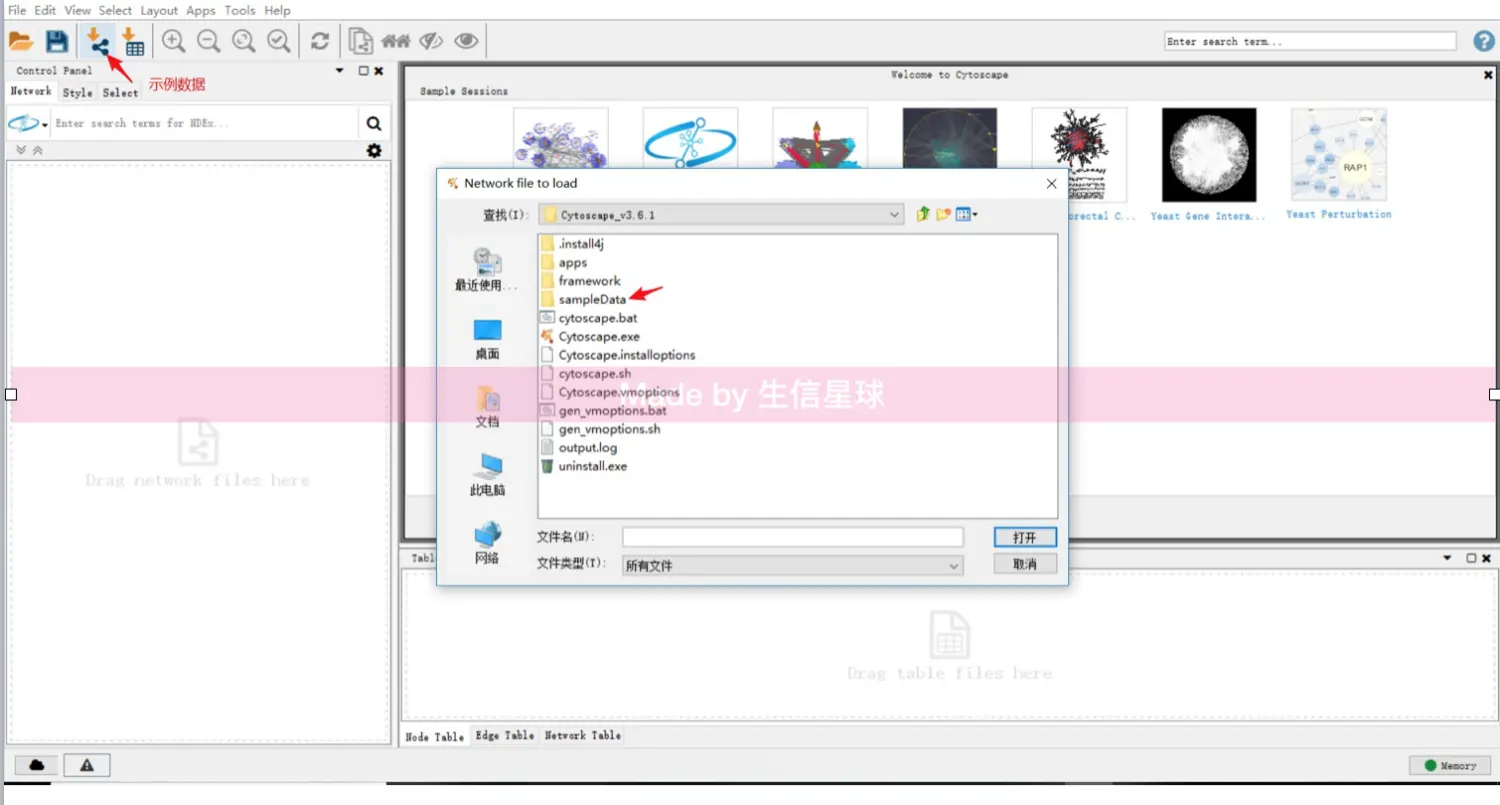
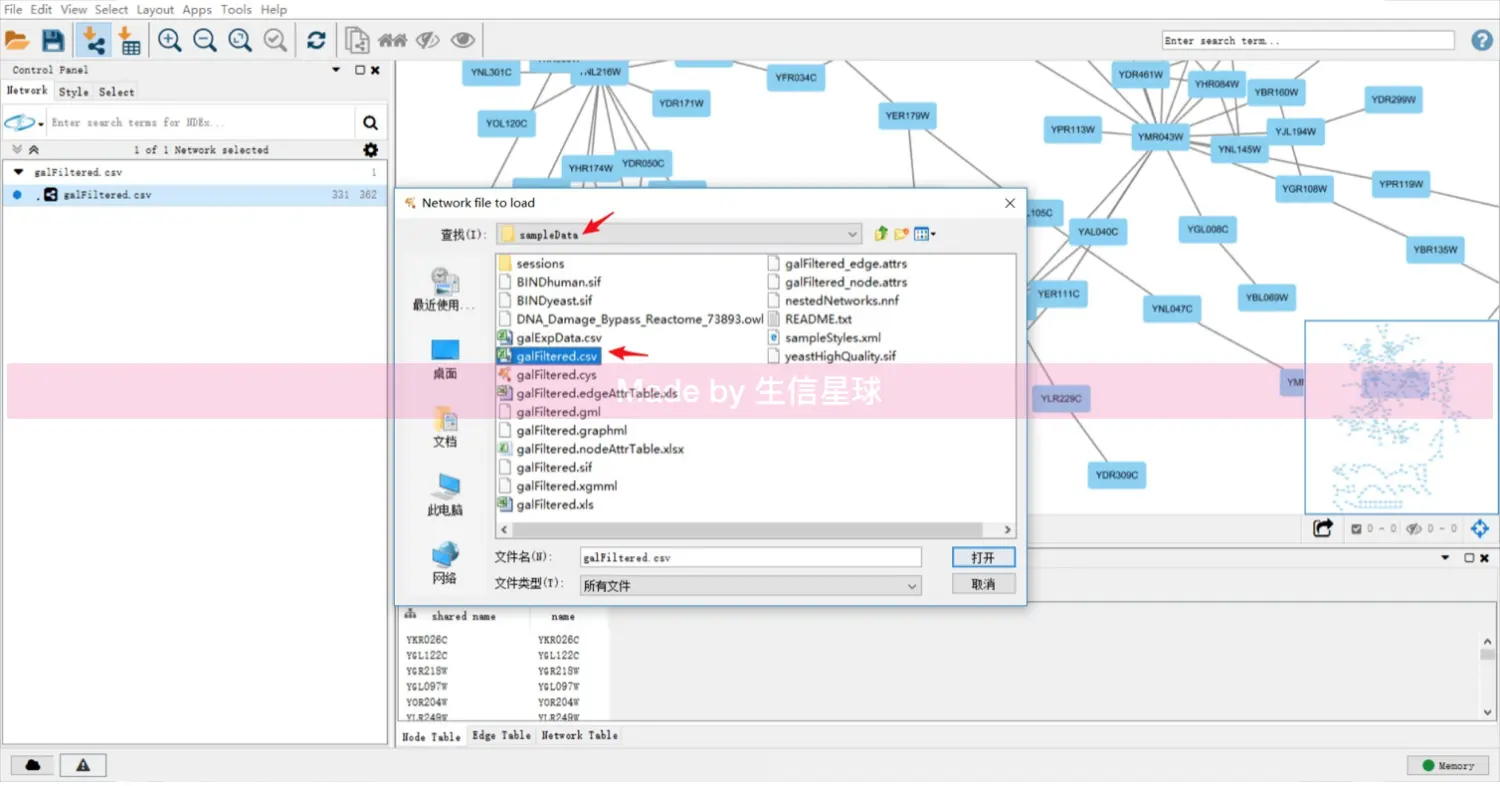
加载数据
输入的node文件和edge文件都是矩阵格式,一般txt格式(除非excel才用xls)
Node设置
颜色,形状,宽,高,以及结点标签
node选择
注意右下方的Table panel,默认是显示所有的nodes。按住
shift然后拖动鼠标可以一次多选许多nodes,同时table panel中的数量也随着你的选择而改变另外,可以直接在Table panel中选择,然后右键
Select nodes from selected rows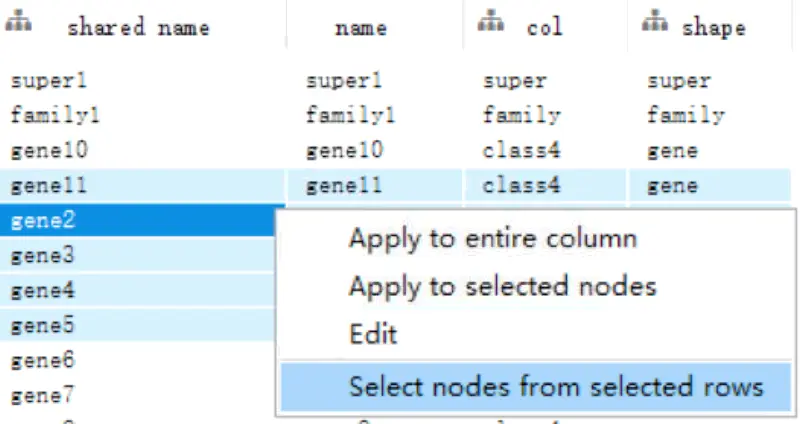
node填充色
选择
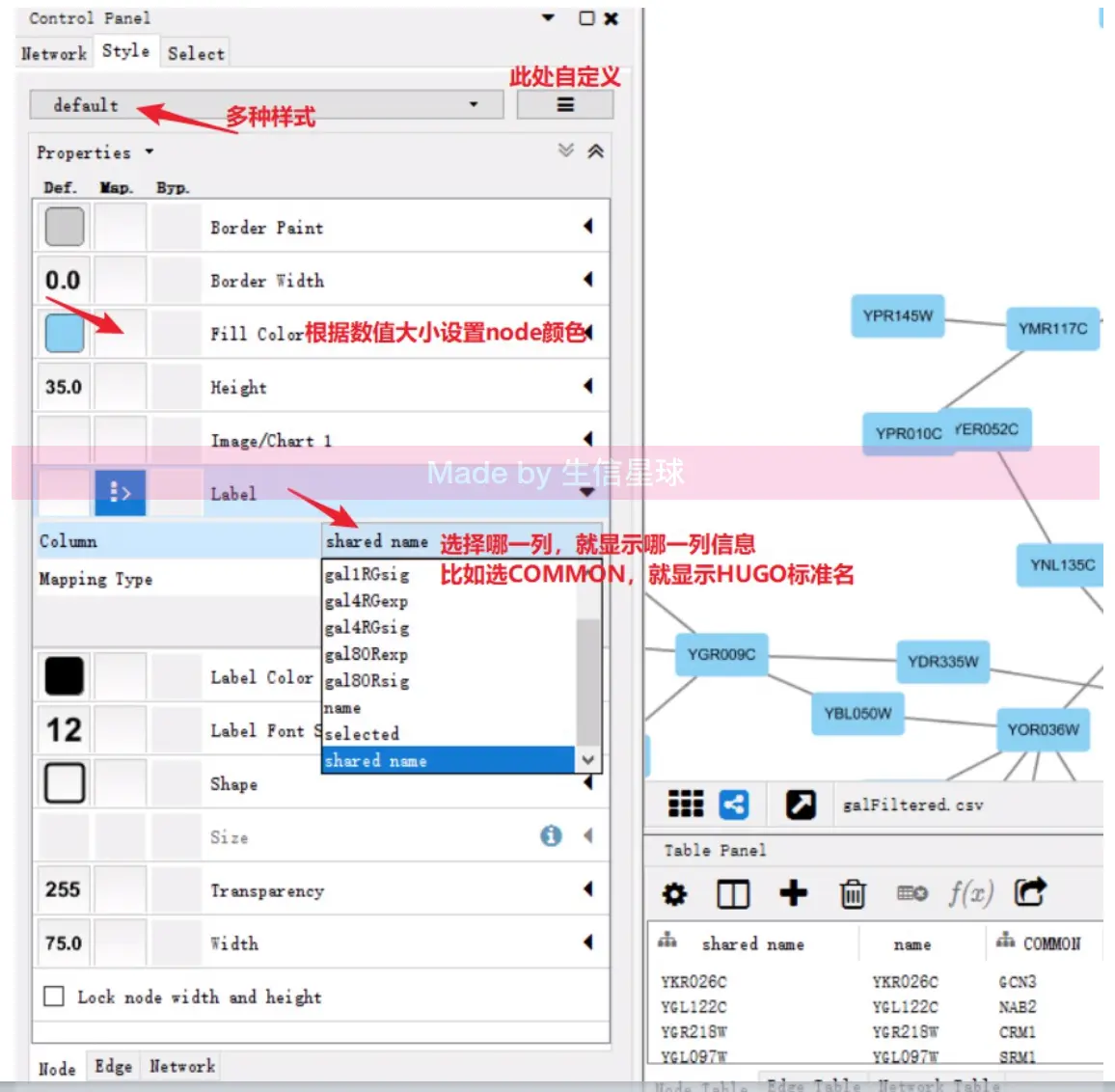
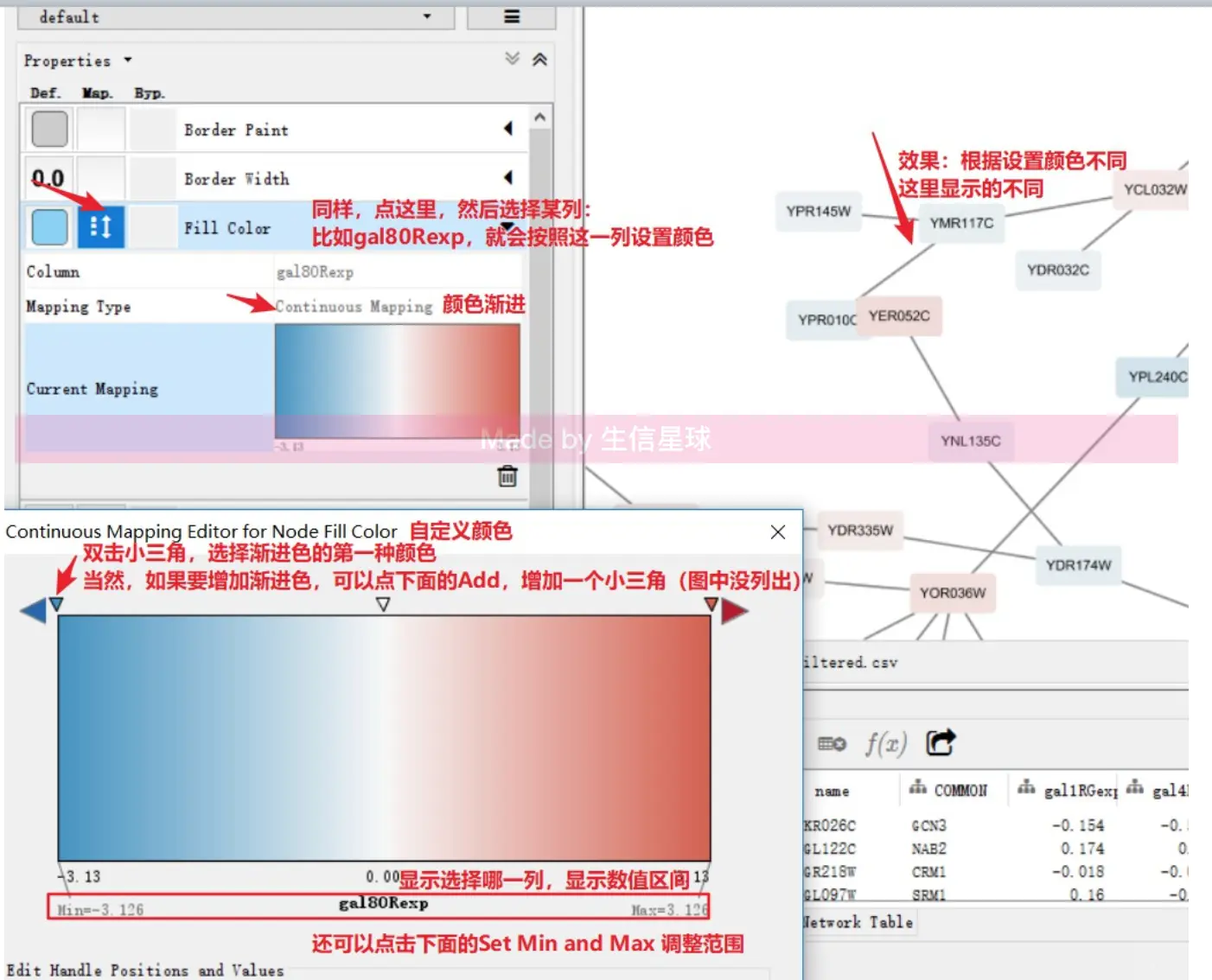
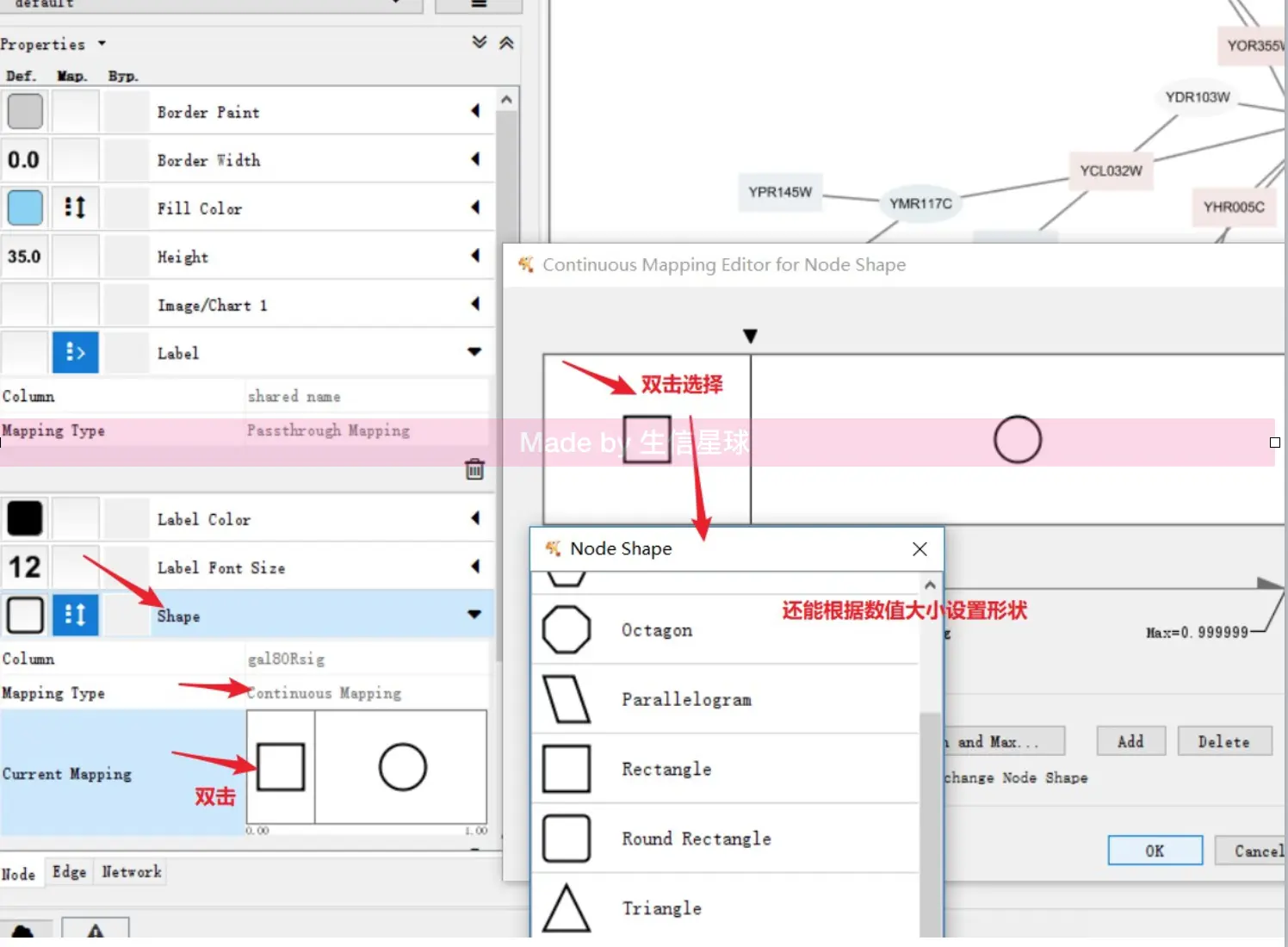
control panel的style,默认进入node的属性设置找到Fill color,点击右侧小黑箭头,会有一个下拉菜单,根据node文件的col列进行设置
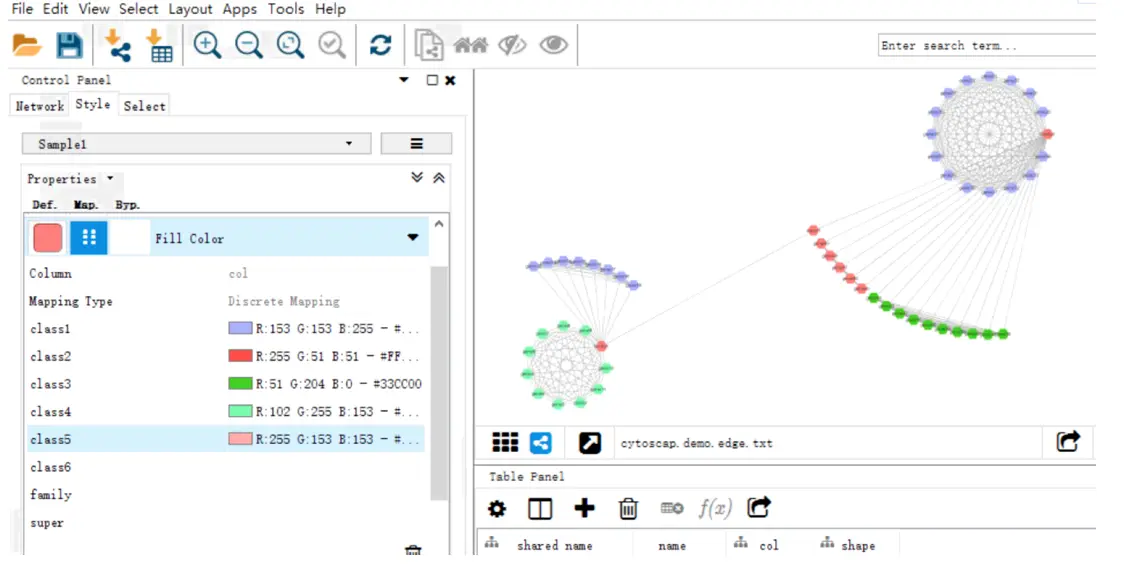
node形状、大小
找到shape,根据node文件shape列进行设置
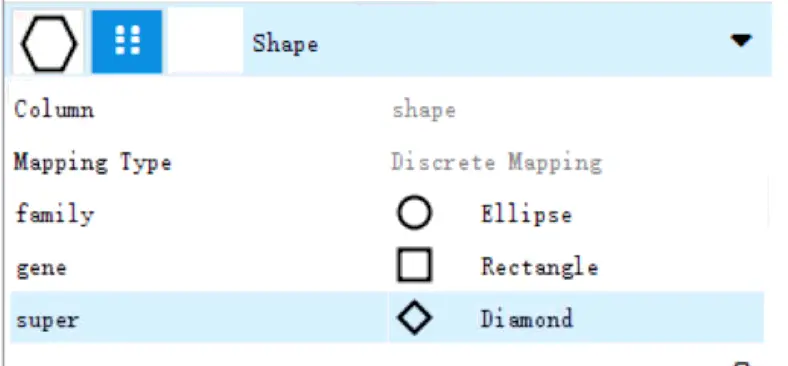
高度(Height)、宽度(Width)、字体大小(Lable font size)都可以设置;另外可以直接用鼠标滚轮放大缩小图片
其他设置
更多的设置在
Properties的旁边小三角中,点击展开,可以添加更多的个性化设置Properties下面三个单词:Def表示默认设置;Map表示全局设置;Byp表示个性化设置
另外可以设置node的排版,在
Layout中有许多样式,比如设置一个Circular Layout就是圆形图显示nodes如果发现有的node排列太紧凑,可以用
Layout=》Node Layout Tools进行整体拉伸缩放、旋转等 ;还能设置nodes的排列方式(排列Align、分布Distribution、堆叠Stack)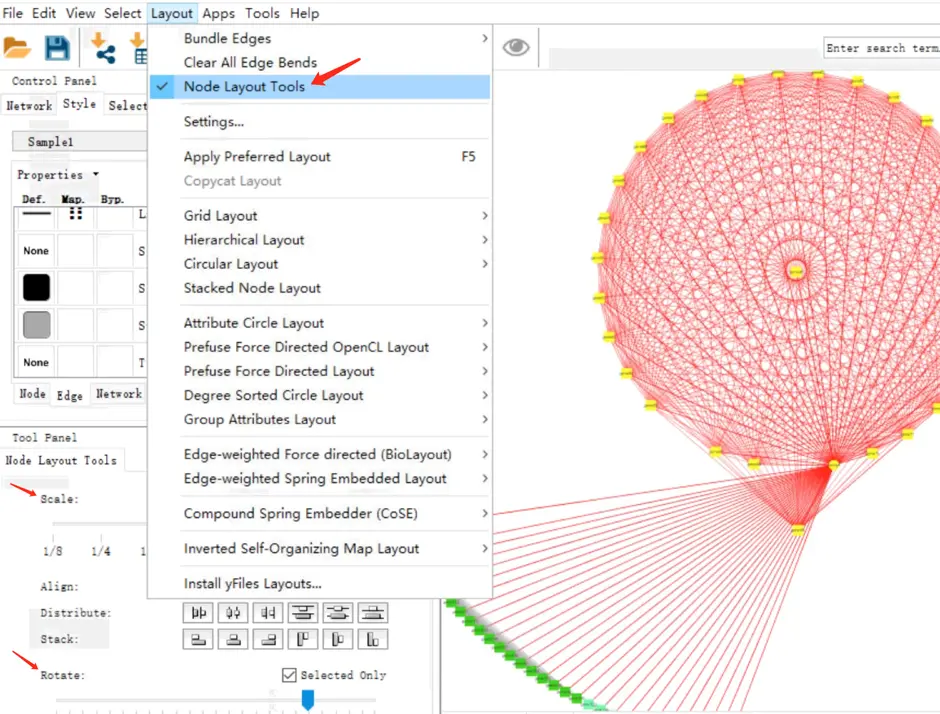
一般不同class的基因放在一起,用同心圆的圆圈一层层排列,组成一个gene family,然后不同的family再直线连接成super gene family。有的时候我们关注一个family内部不同class的不同,就可能会将同一个class中的基因变小;如果关注一个class中的基因,那么就可以适当调大基因的node大小;另外:一个class中基因间的连线除了美观,没有别的作用(不要认为是相关性的连线)
node还能设置边缘框框,可以在其中加图片,加统计图
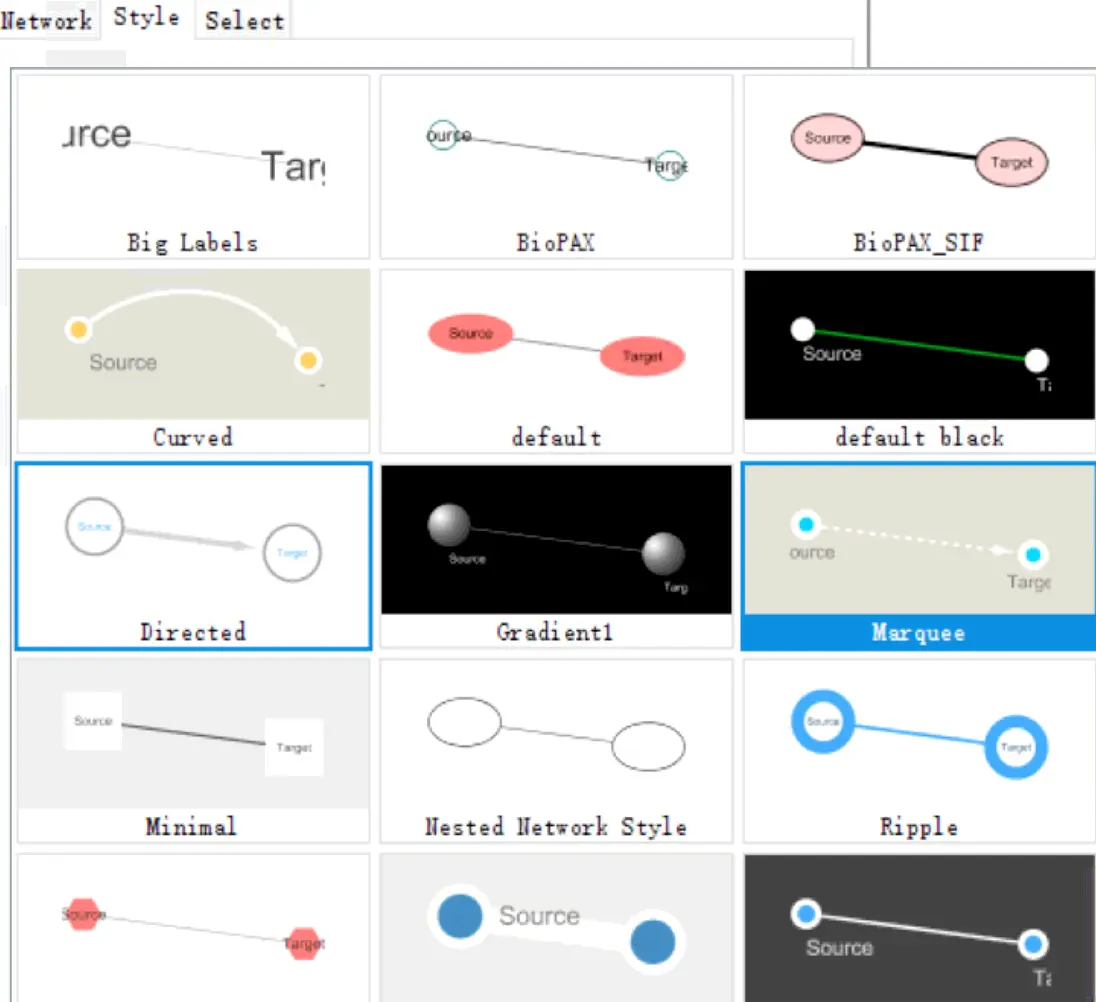
edge设置
主要包括color和width
点击左下角的Edge,可以设置属性
在Style下方有一个长条的选项栏,可以选择许多样式的edge
保存文件
图像一般选择pdf,整个数据集session保存为cys
18.10.13 今晚就学了这些,先说这么多🤔
刘小泽写于18.10.14 跟着cyto导游继续探索网络世界
昨天看了cyto的基本使用,那么你有没有想为什么会开发出来这个工具呢?它美得性价比高吗?值得花费精力去学习吗?用工具和与人打交道一样,初次见面大家都多多关照,但是能否长期交往,还要考察许多个人特征。为了增加对这个工具的信任,我们需要深入了解它一下
为什么要做网络分析?
网络 vs 通路
考察数据之间相互作用,我们应该用的通路类型图更多一些,比如KEGG的pathway分析,二者有何不同?
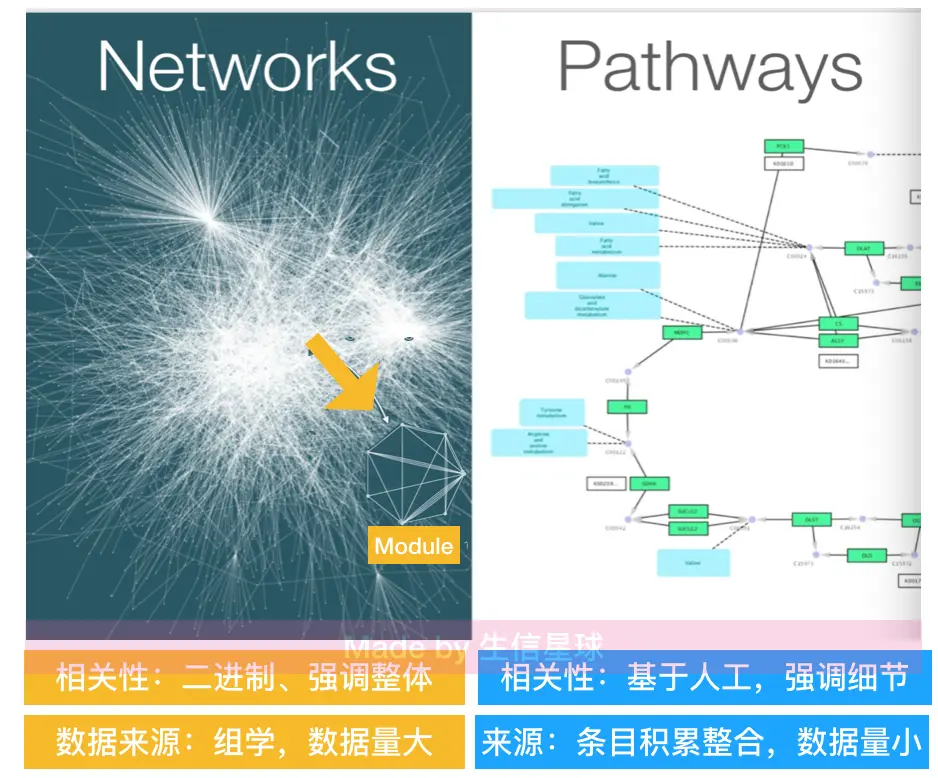
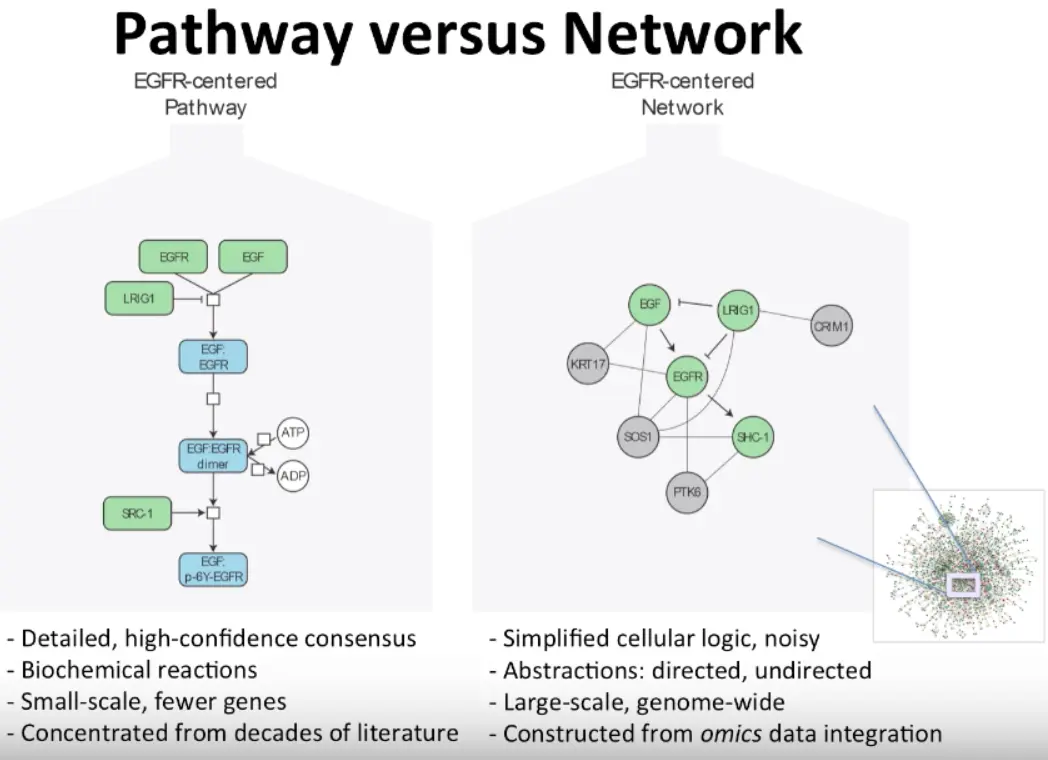
网络分析有几个优势
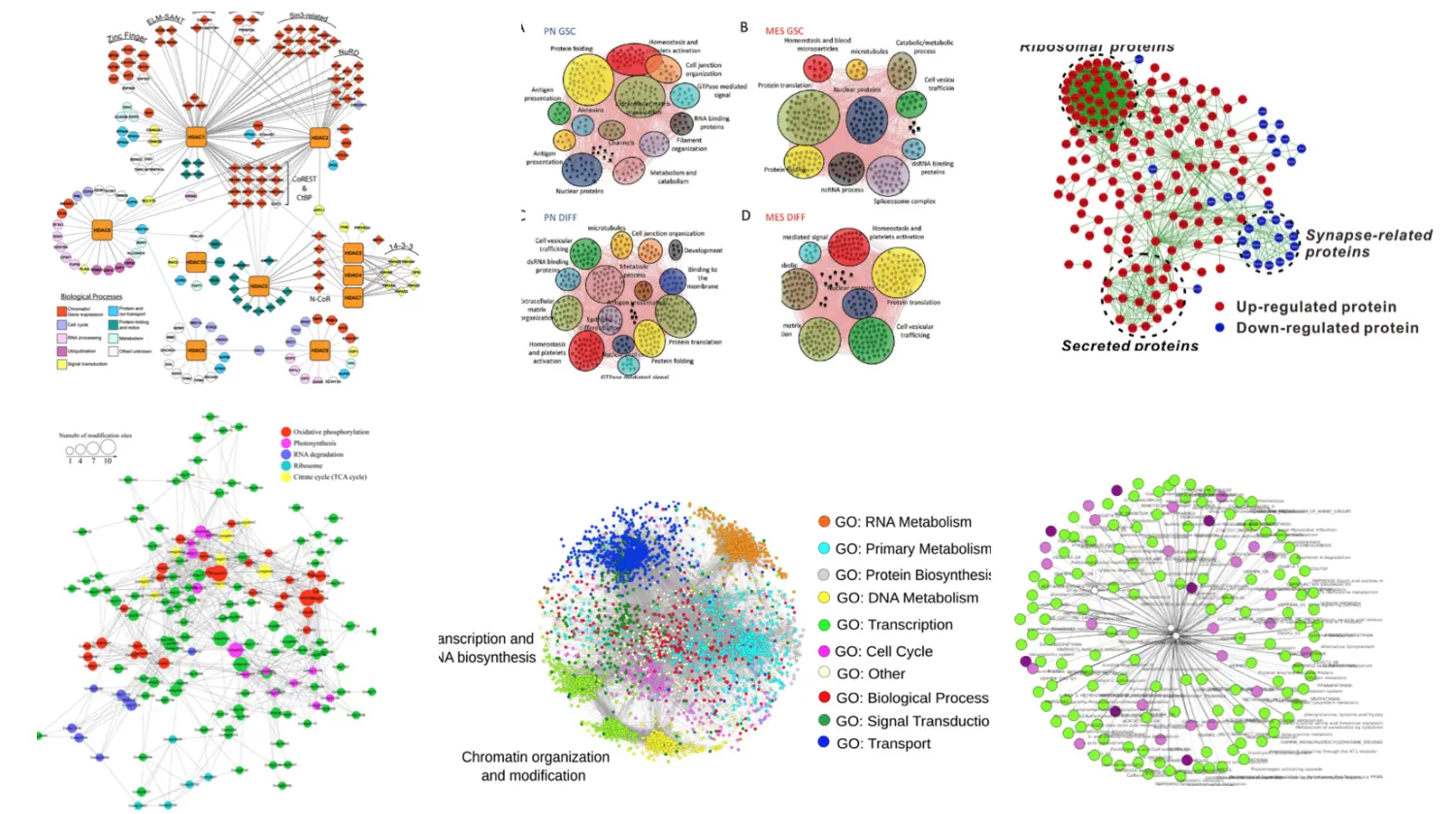
比如你已经筛选了一些基因,现在想看看基因之间的关系,或者它们的功能,就像这样:
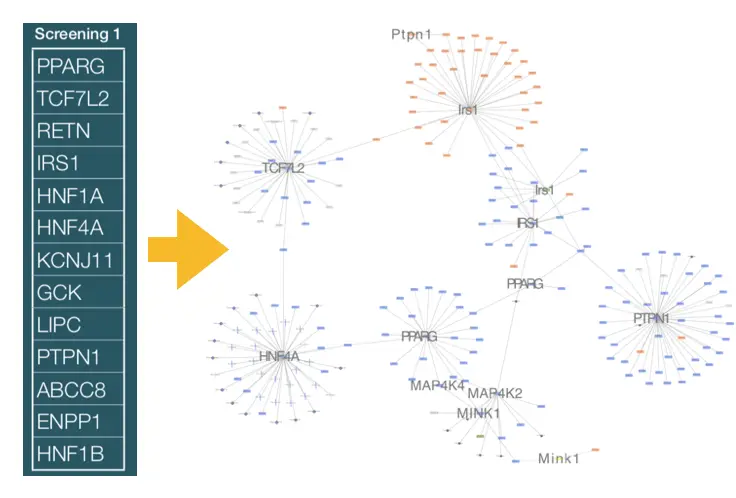
还可以看到组和组之间的联系,比如一个个的gene family,为寻找药物靶标提供了方便
基因之间的联系可能并不都是直白的两两联系,还有就是只有基因的某个因子和另一个基因的因子有关联,这个在通路图上可能显示的就不是那么清楚。而在网络图中,只要这个点我们感兴趣,就能给它坐标,接下来就是点到线的过程了。当然根据算法不同,点线之间的连接方式也不同
其实工具不重要,重要的是数据,这句话放到哪里都有用 网络可视化工具:
一般会有两种人需要需要用cytoscape:一种是想省事,不用编程就能做;另一种是需要发文章的,用R做的网络图不好精修,于是用这个。就像许多R做的图到了发表级别都需要AI处理一样
为什么这么多人选Cytoscape?
我们从不缺少美,也不缺少发现美的眼睛,这个网站就列出了许多的可视化工具。问题就在于:如何选择合适自己的,选择后是否能认真学下来 http://selection.datavisualization.ch
Learning Tools = Saving Your Time
看文章引用量破万就能知道,这的确是一款神器,在生物网络拓扑构建领域中,cytoscape可以说是领先的。but why?
首先,它支持的网络种类很多。比如蛋白互作(PPI)、转录调控网络图(TF-target)、网络聚类模块分析(Module)、miRNA调控靶标基因网络图、竞争性内源RNA网络(ceRNA)、通路交互网络(pathway-crosstalk)
其次,它灵活多变(用熟了是灵活,否则就是臃肿的参数设置),可以随意调节节点与变线的各种属性(如颜色、大小、粗细、形状等),保存格式多(最常用就是pdf),可以保存session方便以后修改(和R差不多),**各种实用的插件(**clueGO+cluepedia、bingo、cykeggparser、metdisease、mcode、cytoHubba、iRegulon、enrichment map等)Cytoscape的APP Store在http://apps.cytoscape.org/
给不同的人提供多种选择,大家都喜欢可视化,但不是所有人都喜欢图形界面,因为那里存在着太多的参数设置让人头疼,因此Github上Max Franz等人还为程序猿提供了网络可视化的js版本http://js.cytoscape.org/【今天偶然间发现的,惊喜😋,因为以后可能不用安装桌面软件就能进行网络可视化了,我个人还是更期待用程序去设定而不是用鼠标】
画图有许多工具,比如AI、PS也能画出网络图(注意是画!)使用专业的绘图工具,你需要指定每一个节点的颜色、每条线的特性,它是像素决定图形
所有的细节你都需要考虑,这对于一个数据分析者来说无疑是最头疼的 这也是我开始为什么犹豫学习这个工具的原因,因为不想浪费大量的时间在图形处理上。于是我也能理解当时jimmy使用这个工具的心情了(http://www.bio-info-trainee.com/2055.html)

而用cytoscape恰恰相反,你的数据决定图形,只要能理解需要提交的数据意义,做的图就是问题不大的
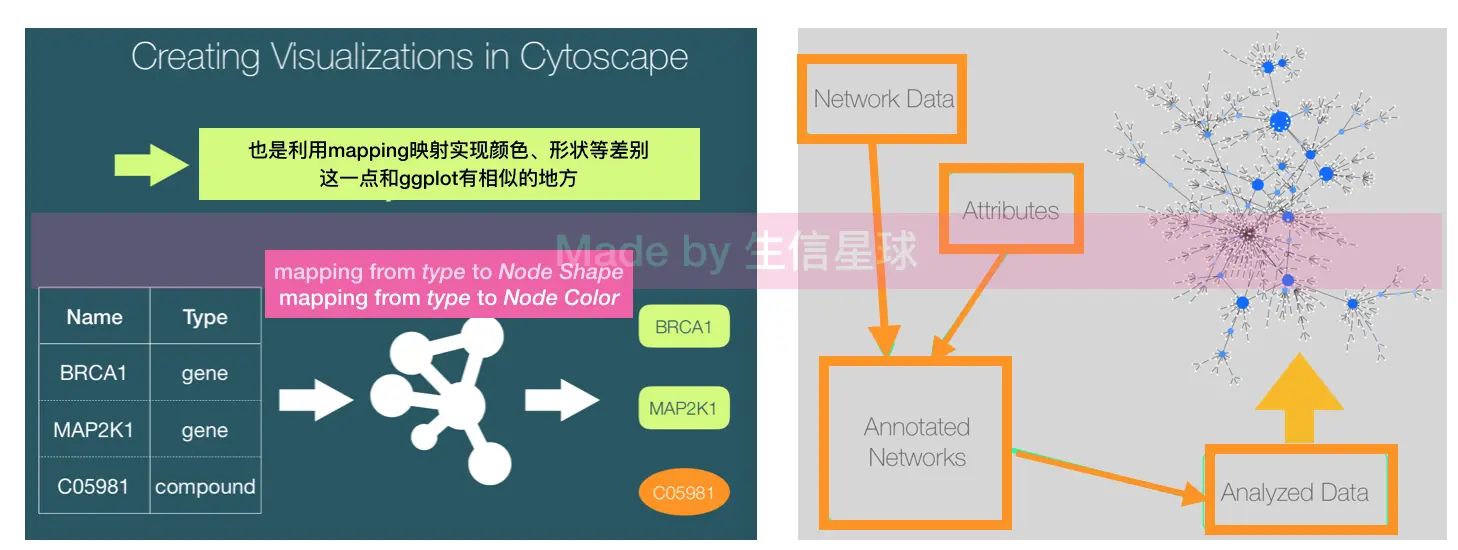
绘制网络图注意问题
避免加载太多数据,尤其属性太多会使图形很臃肿;另外多个数据集放一起,很难观察总体的趋势
一般给node上色是为了突出不同的基因和因子,以及它们表达量的差别。不要为了美观再给edge/label上色,那样会很杂乱
如果要画其他基因相关数据,不要让它们与主要基因的图重叠,可以是使用线的粗细、虚实来区分
Cytoscape与Cytoscape.js
它们长得像却是两款独立的软件包,不过这并不妨碍它们做相同的事,那么它们有什么异同呢?选择哪个更适合自己呢?
| Cytoscape | Cytoscape.js |
|---|---|
| 桌面级:基于java软件 | 网站级:基于java script库 |
| 需要PC运行 | 支持大多浏览器(包括平板、手机上的) |
| 必须安装Java | 没有要求,只需要浏览器 |
| 鼠标点点点 | 键盘敲敲敲 |
| 用App拓展 http://apps.cytoscape.org/ | 用Extrensions拓展 http://cytoscape.github.io/cytoscape.js/#extensions |
| 数据映射:设置Style | 数据映射:设置基于CSS的Style |
不过这二者未来有可能会高度整合,因为目前cytoscape支持了Cytoscape.js network/table JSON (JavaScript Object Notation)文件的读写,另外cytoscape可以将style转成Cytoscape.js的style格式。
上面说这么多,都是为了加深对陌生事物的理解,和它混熟了学起来才更有效
网络分析的一般步骤
1 创建网络
R或者Excel
2 载入数据
2.1 数据格式
Cytoscape支持多种数据文件格式,其中主要的有3类:
sif格式:
nodeA<interaction>nodeB nodeC<interaction>nodeD …包含3列,1和3列是有互作的基因名或蛋白质名等,第2列是相互作用的名称
xgmml格式:xml格式,可以规定节点和边的许多信息,比较复杂
txt格式:用tab分割的纯文本文件
至少两列,每一列都是基因名(或蛋白质名),同一行的两个基因(或蛋白质等)代表有互作关系,比如两列gene id 可以追加参数,比如设成3列:加上两基因调控的强弱系数 可以四列:例如是否共表达(1、-1表示)
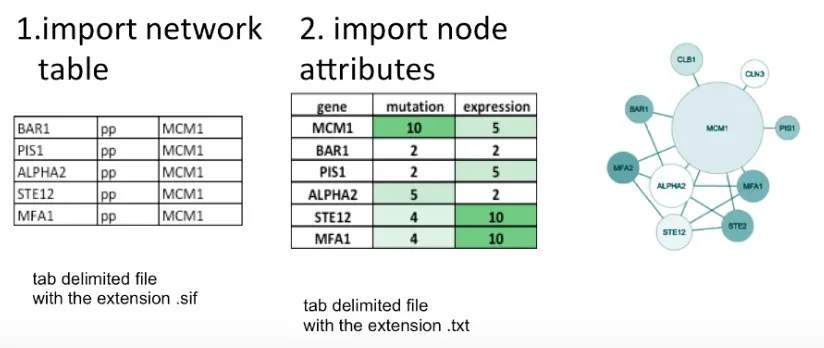
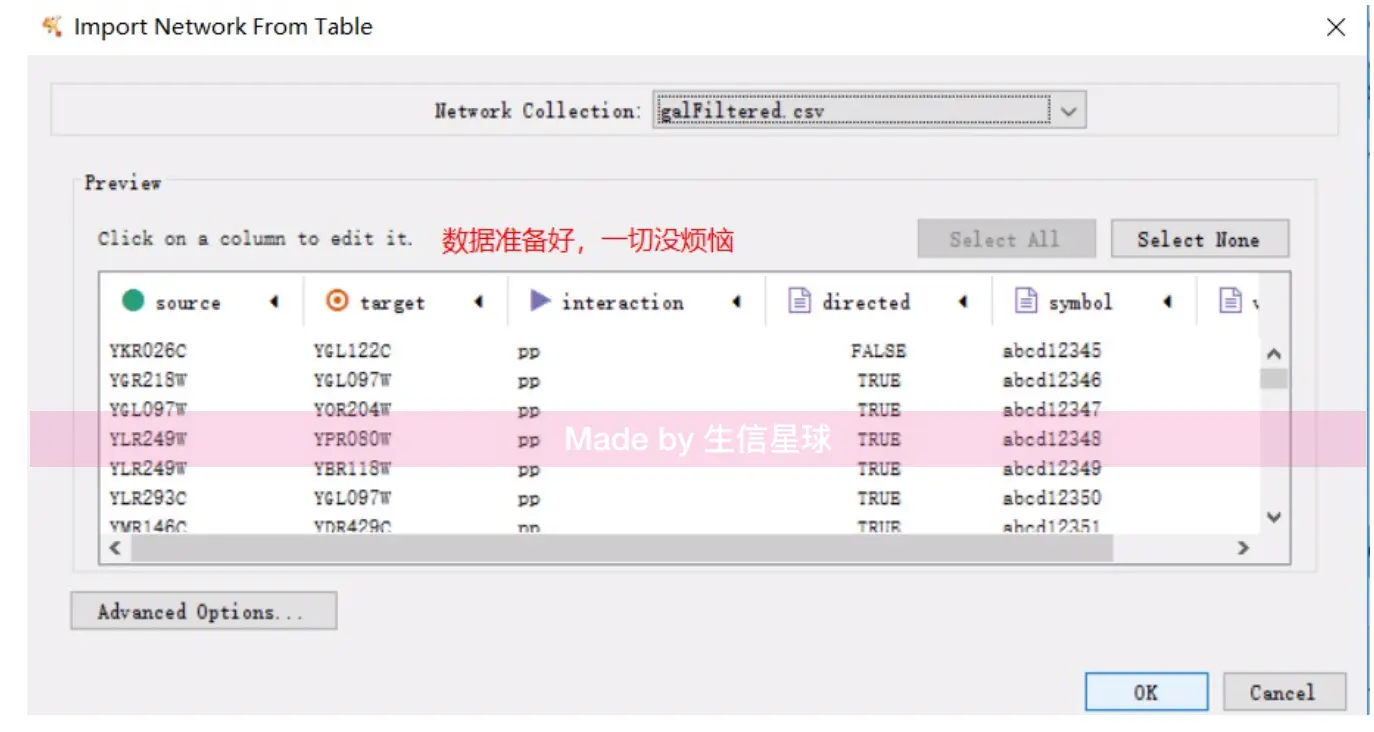
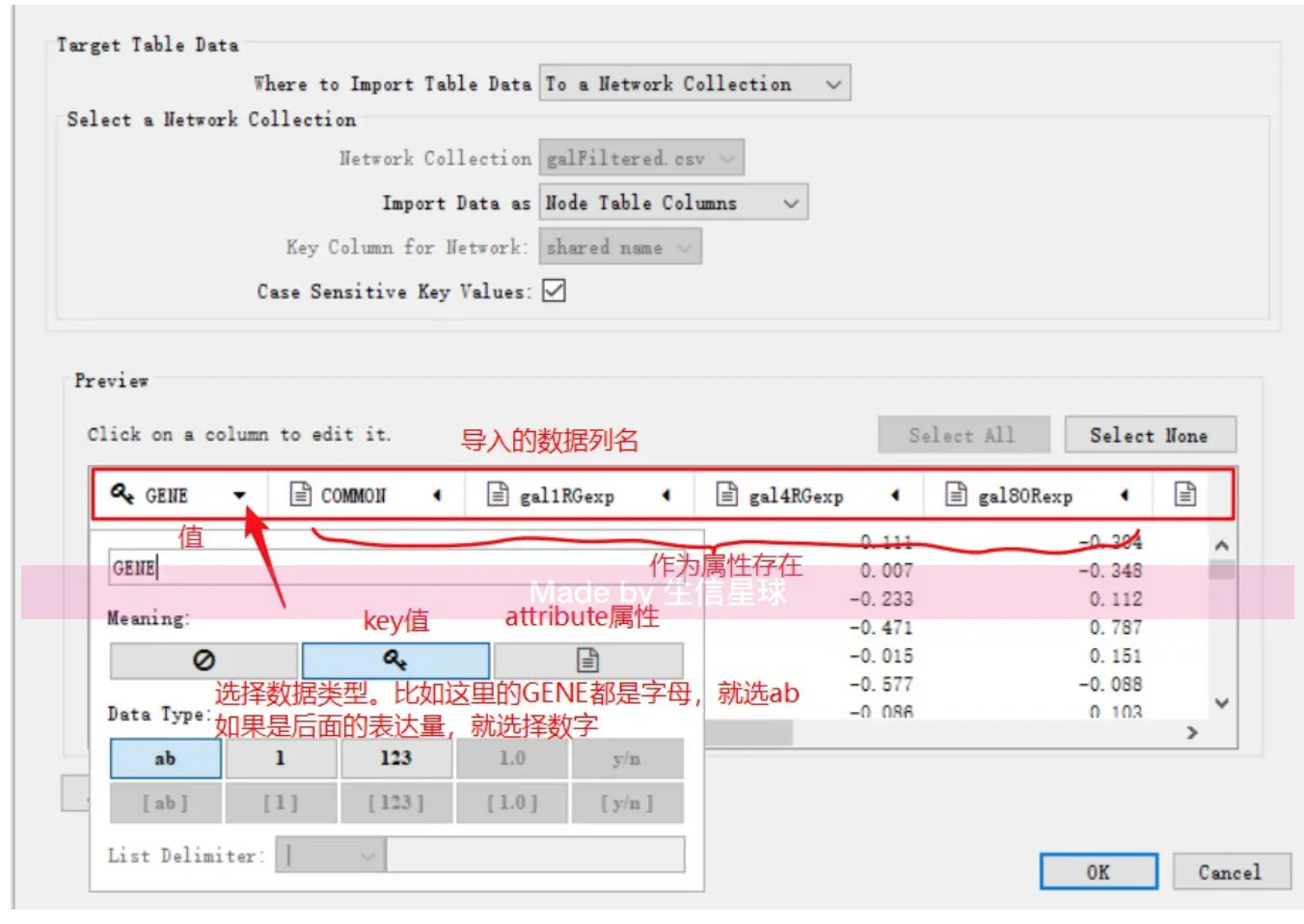
导入数据文件
network table(以.sif格式为例):上图中第一列是自己实验中的基因数据,第三列是分析得到的相关基因数据,例如:实验中BAR1基因与MCM1基因是相关的,也就是说,它们之间一会是要连接起来的再导入节点属性文件
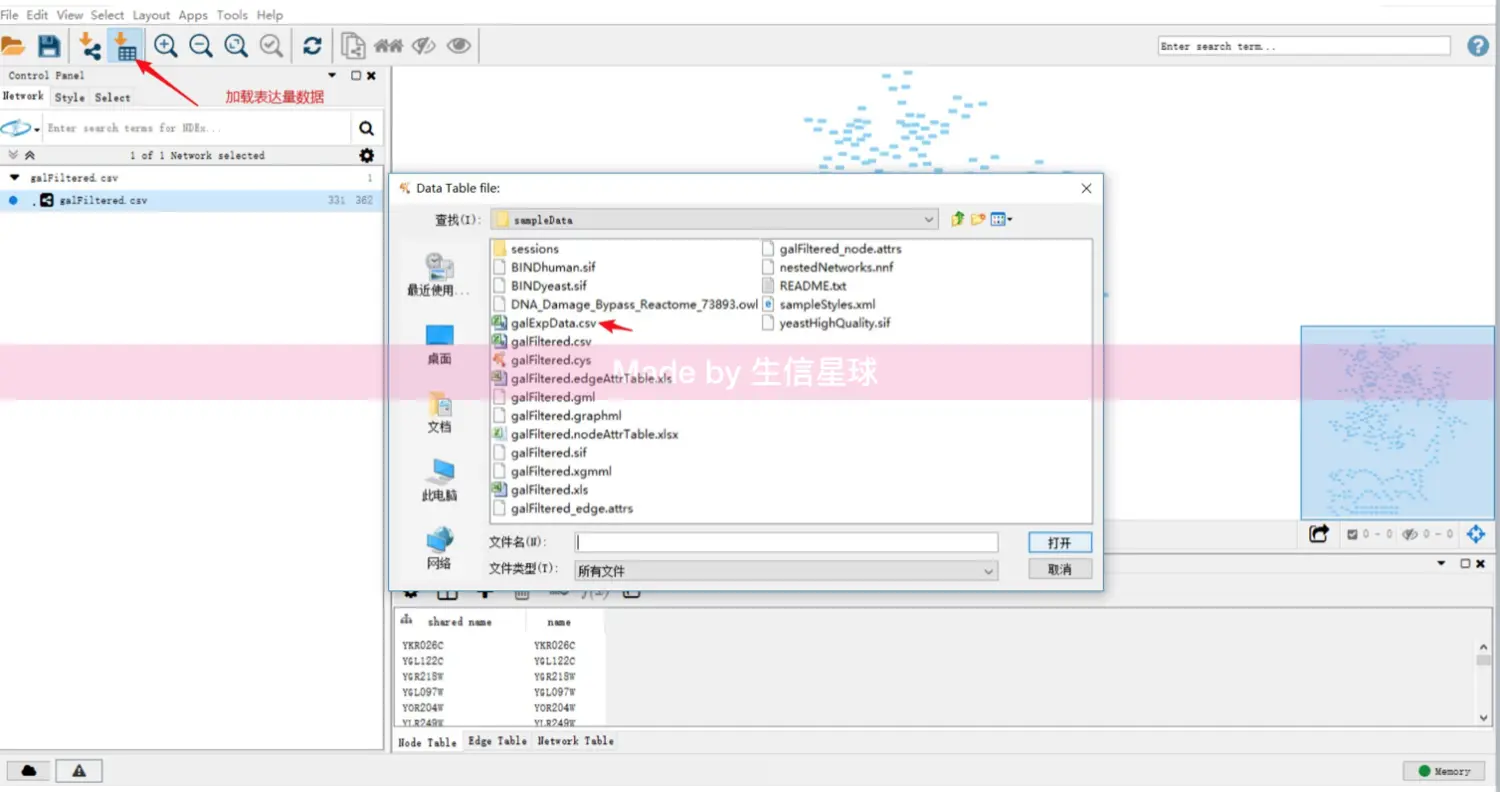
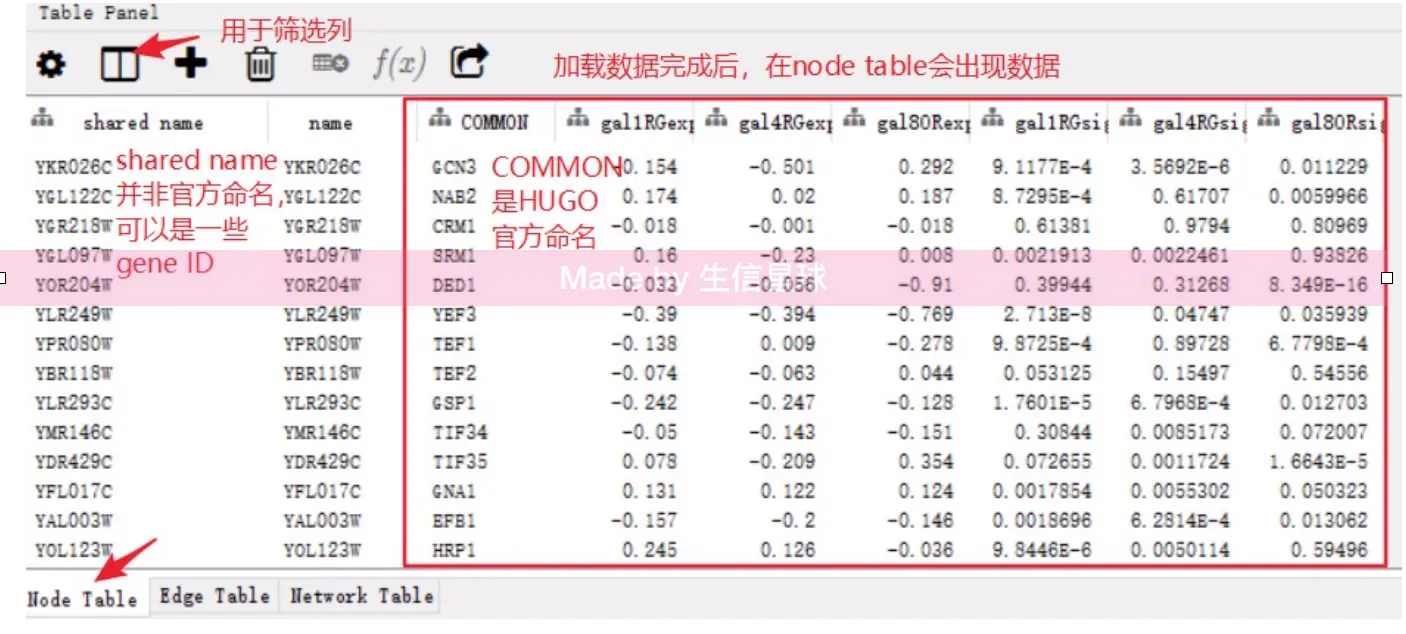
node attributes(.txt格式): 第一列是gene id(与先前导入的数据文件的gene id保持一致),后面可以自己设置(这里增加了mutaion和expression)
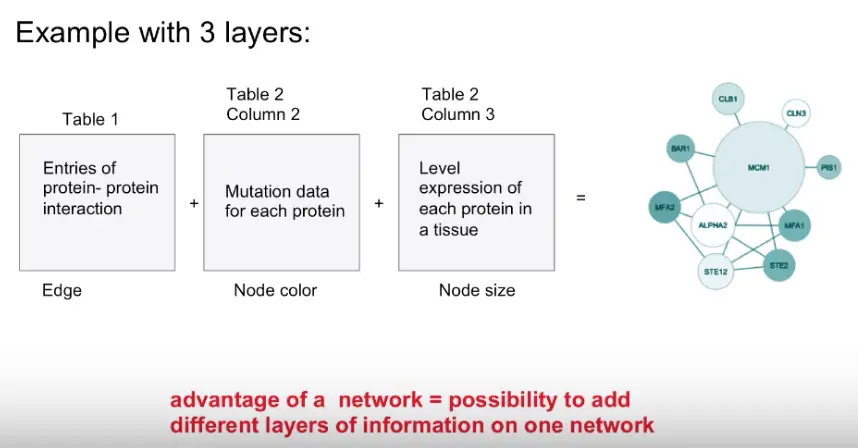
2.2 新网络雏形
之前导入的第一个数据文件在网络图中作为线存在,为连接nodes;
导入的节点属性文件中第二列的mutation信息,作为node color;
节点属性文件中第三列expression信息,作为node size
…而这些都是添加进来数据的时候,自己设置好的
因此可以看出,只要提供不同的属性信息,就能在网络图中显示不同的样式 虽说它不像ggplot是图层叠加,但是它的原理也是一环套一环,生成的图层是相关联的
3 新网络设置
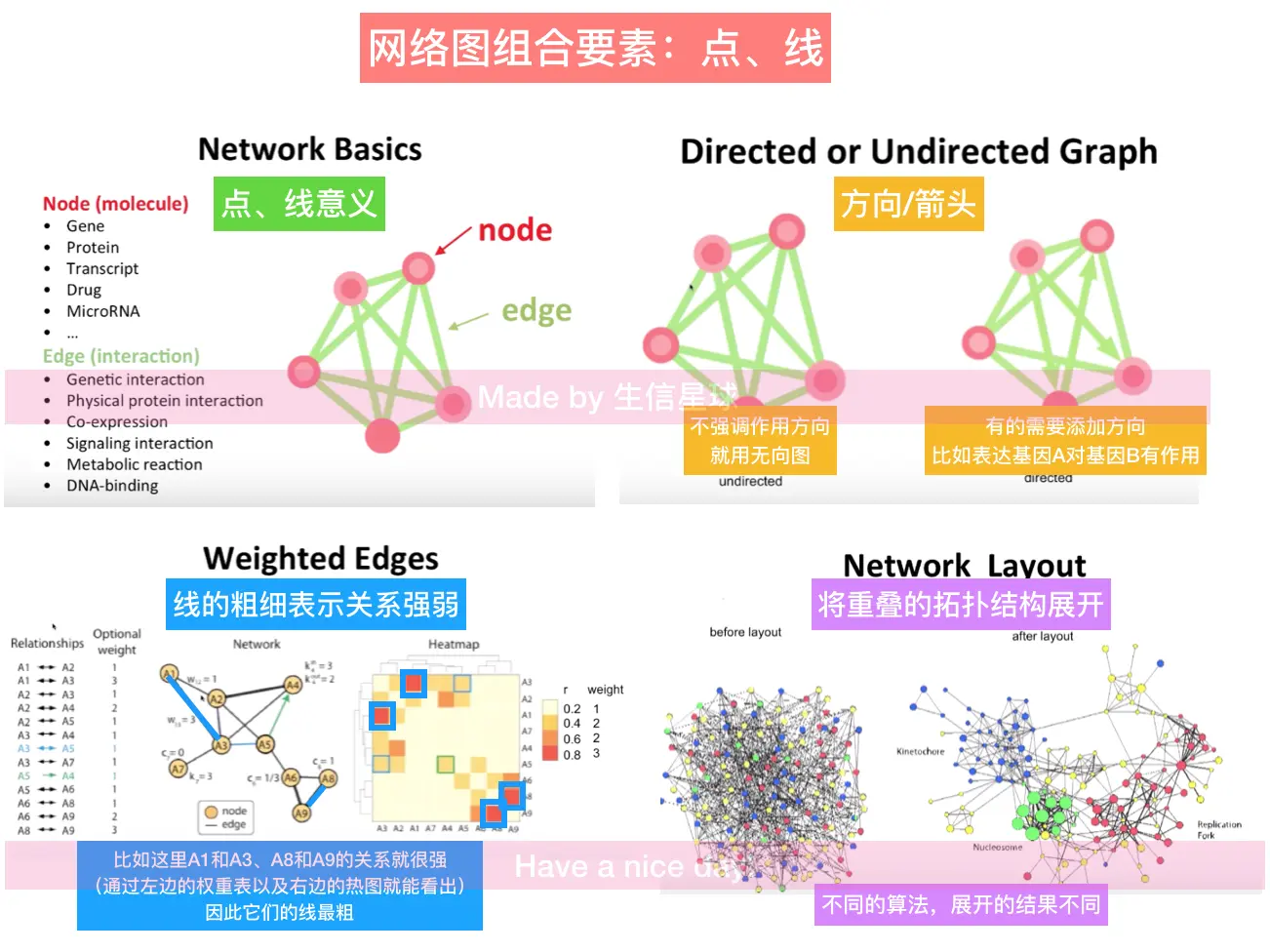
新网络的要素:点node、线edge【点又可以通过颜色、大小分区;线可以用方向、粗细区分】原理就是下面👇的图,具体操作昨天应该提到了
4.1 利用app- app分类
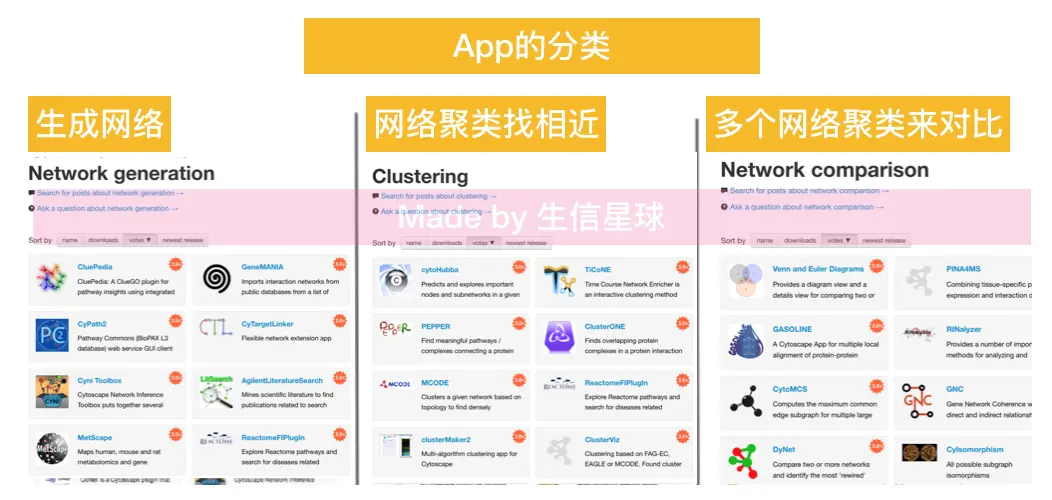
看能得到什么信息,是不是有意义的网络图,利用Cytoscape的特色:Apps
数量巨大,但主要分为以下几类
4.2 利用app-比较好用的一些App
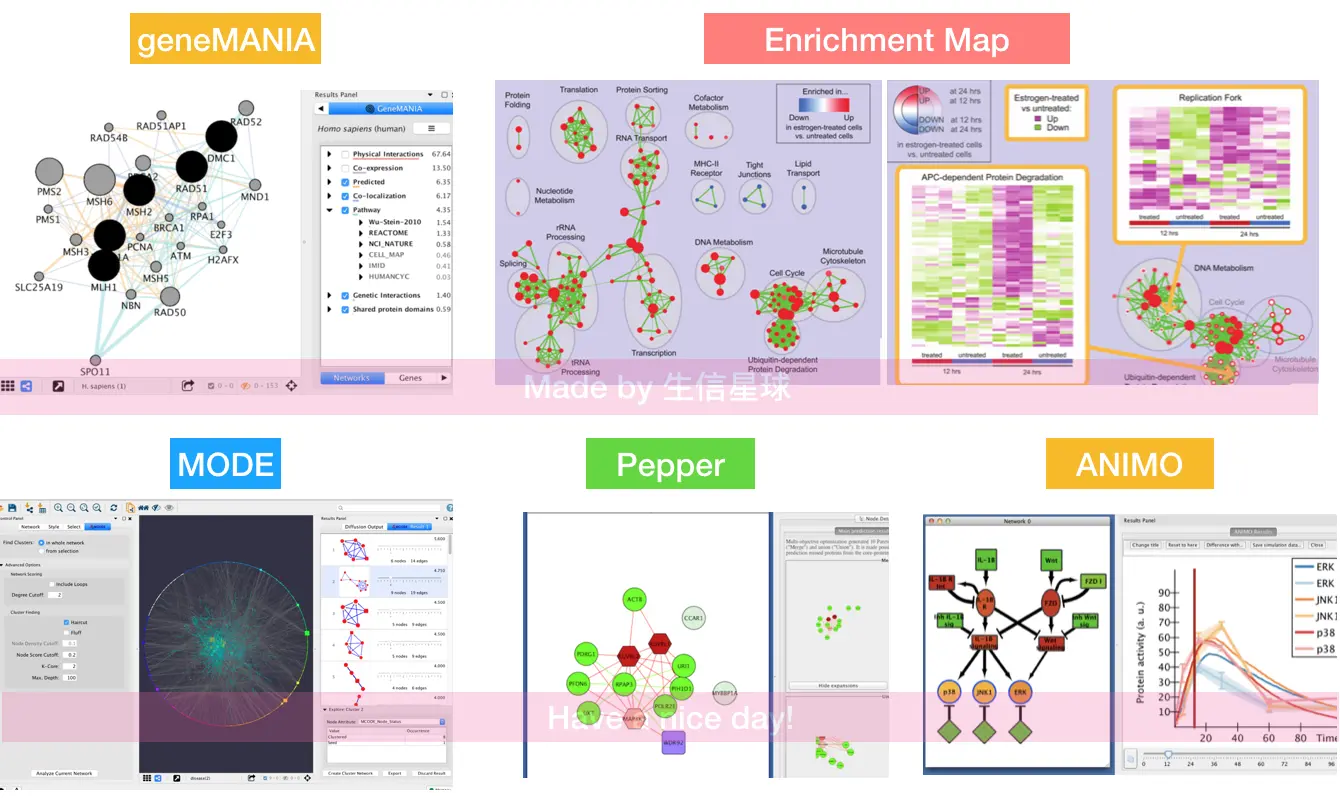
geneMANIA:基因功能预测,它会把相同生物过程的基因集或蛋白组合在一起,然后看要研究基因和它们的关系。比如:你可能不认识豆豆,不知道豆豆干嘛的,但是你可能认识花花,然后发现花花每天努力学生信,发推送,然后猜测:嗯,豆豆可能也是干这个的【傍晚写到这里,突然想花花了🤡,周末两天培训班,结束了快回来吧,还等着共进(食堂)晚餐呢】
McodeMolecular COmplex Detection:检测蛋白互作网络(PPI)中密切相关的区域(可能表示分子复合体complex)【相比于spring(最多提取2000个nodes)提取的信息更多】,也可以检测motifs、feedback loops
ANIMO**(Analysis of Networks with Interactive MOdeling)**: 建模挖掘通路,对生物通路建模,与湿实验数据动态比较。例如,想要理解某个分子变化对通路的影响,就可以用这个
Pepper:预测新的互作关系,还能根据结构和功能进行蛋白的排序,除了按照连接特征(拓扑系数)外,还整合外部数据库(GO注释,比对已知的复合体)
Enrichment Map:基于网络的基因集富集(支持GSEA数据),将基因集组合成一个网络(cluster),nodes表示基因集,edge表示基因成员的重叠,node颜色表示富集程度。其中的热图可以让用户探索富集基因集的细节
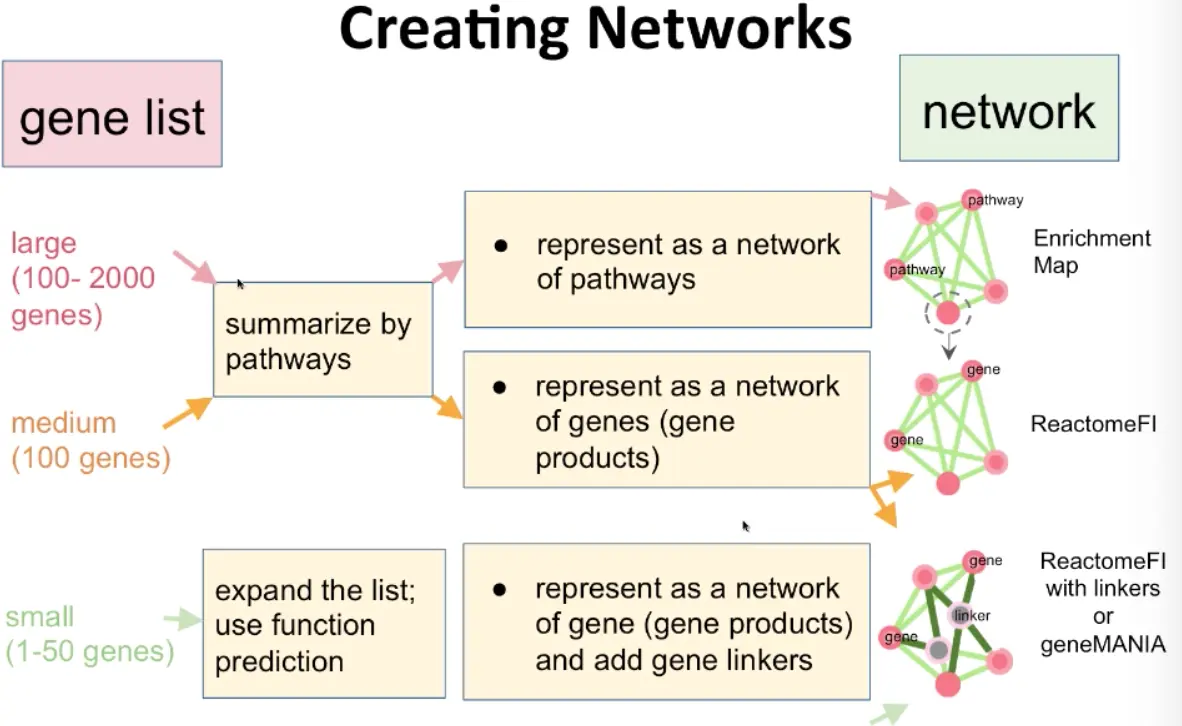
4.3 根据基因列表大小选择不同的app
大基因集(100-2000基因)=》利用通路缩小范围=〉展示成通路网络=》可以用EnrichmentMap
中等基因集(100基因左右)=〉利用通路缩小范围=》展示成基因/表达产物网络=〉可以用ReactomeFI
小基因集(1-50个基因)=》利用功能预测扩展范围=〉展示成基因/表达产物网络,并且增加基因linker=》可以用带有linker的ReactomeFI或geneMANIA
5 导出网络
(table格式)以及图片(pdf等格式 )
简单的演示