010-认识高性能计算HPC
超算集群系统的组成
当前HPC的主要架构包括集群和MPP(大规模并行处理)两种
集群架构是目前最广泛的超级计算机架构。
典型的HPC集群系统主要由六类计算设备和三类网络组成。
六类设备
主要是指管理节点及登录节点、计算节点、交换设备、I/O设备和存储设备。
登录节点 登录节点相当于用户访问集群系统的网关。用户通常登录到此节点上编译并提交作业。它是外部访问集群系统强大计算或存储能力的唯一入口,是整个系统的关键点。为了保证用户节点的高可用性,一般采用硬件冗余的容错方法,如采用双机热备份的方式,或者采用RAID(独立磁盘冗余阵列)技术保证用户节点的数据安全性。 登录节点一般对计算性能要求不高,整套集群根据需求配置数台机架式服务器即可。
插播一个很尴尬的三段式小故(tu)事(槽): 【第一段】之前我有一个HPC账号,具体配置如下,看着很高大上

兴高采烈登陆上去,先查一下内存吧free -h,结果返回来总共64G,剩余33G;htop一看线程数才12!怎么回事!我蒙了,这确定是集群这么强大的服务器的配置吗?和配置清单不符!还没有我其他的服务器好用呢!试着跑几个程序【核心数设为8,不敢设多】,结果没过几分钟就挂了,试了好多次没有成功过~一气之下将它抛弃,还是用了我用着顺手,就是硬盘空间只有可怜的100G的服务器… 【第二段】故事还没完~后来意外得到一台强大的服务器,内存500G,硬盘10T,cup线程64,把我开心的啊,真的是欣喜若狂。因为你可以想象一下跑一个程序为了省点空间,把文件解压完又压缩,用时再解压,很崩溃😵这回好了,10T空间,豆豆再也不用担心硬盘了。后来就发现,原来强者的背后还是孤独和寂寞—没有连外网(意味着我只能通过内网访问,也不能直接下载软件和数据),也没有专业维护(意味着后期会出各种各样的问题)。果然,当我传完200多G的软件和数据后,准备一展身手,这时由于突然停电,服务器没有UPS给服务器续命,强者服务器喷了一口鲜血倒地~后来硬盘坏了,数据也没了,忘了猴年马月修好的,反正过了很久很久,那期间我还是继续用我的乞丐机服务器【现在看来的确是的】。后来不知机房老师心情好了还是喝多了,重装了系统,清空了一切,我想,从今以后我一定要及时备份!只要有那10T硬盘,我还是愿意重新来过的~满怀虔诚地鼓捣着服务器,好景不长,现在它又倒地了~而伪服务器管理员自己用服务器满64线程跑完程序后,不管了,强者又受重伤,这次最惨的是没药,救不好了
【第三段】嗨,生活嘛,本来就充满了许多的不如意~我并不会在意🙃😠。继续用我的乞丐机,一直在学不用计算资源的R。直到昨天晚上,手有点痒,想敲一敲键盘,意外的想起了我那失联已久的HPC账号,看看能不用吧,起码硬盘是管够的【没记错的话,剩余硬盘空间还有500T】。 这一次,我毕竟有使用两个服务器的经验了,再用一个问题应该不大。我注意到当时发来账号的时候,还附带了使用说明,我以为和普通的服务器一样就没管。以前看着还挺麻烦,还看不懂。昨晚我仔细又研究了一下,才发现了其中的奥妙!
原来HPC不能按服务器来用,最大的差别就是节点问题 服务器没有什么登陆节点、计算节点之分,登上去就能用; 而HPC呢,就像这篇文章介绍的一样,他有许多的节点,你用ssh登陆进去的看上去像服务器的,那是登陆节点,专门为用户登陆准备的【话说一个登陆节点就要用好几台小服务器,是不是有点太奢侈!】 如果要运行程序的话,需要用计算节点。我又重新看了看人家“真”管理员发给我的使用说明,我登陆进login2级节点,用qnode看了下,瞬间整个人都呆了

原来我囚禁了一头猛兽这么久?

而这仅仅列出了15个节点而已!一共有60个计算节点!
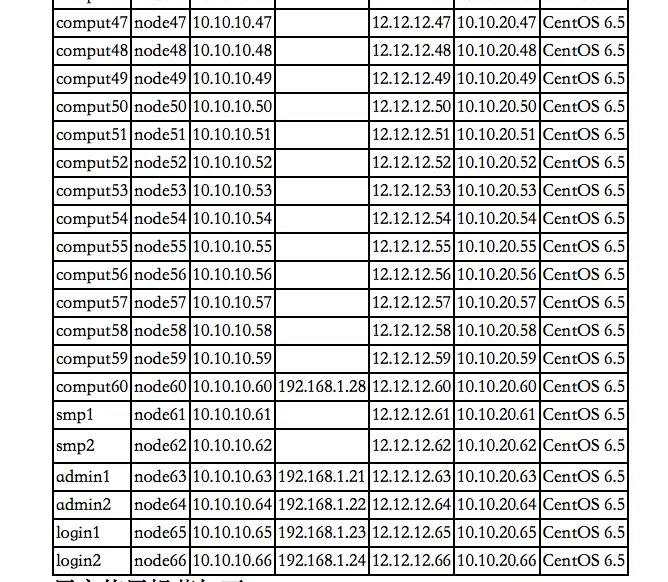
好吧,这回清楚了,login1和2是计算节点,也就是之前以为是计算用的64G内存的那个界面。60个计算节点,一般都是20核、128G内存,还有几个256G内存的,这么算下来,所有计算资源算上,内存得有1T+,cpu核心数1000+【对比下普通笔记本:8G/16G内存+4/8核cpu】 很刺激有没有,我为猛兽接触了封印,拉出来溜溜先
管理节点 管理节点是集群系统各种管理措施的控制节点。
计算节点 整个集群的计算核心。分为胖节点(多路,比如这里的两台8路大内存服务器)和瘦节点(双路)。
异构节点 同时使用CPU、GPU或MIC,可以大幅度提升计算效率。
交换设备 集群各节点之间需要通过网络连接在一起。
I/O设备和存储设备 存储数据和提高读/写带宽。
三类网络
- 管理网络 用户管理节点和各计算节点、I/O节点的互连,所连接的机器就是集群内部的本地机器,所以不需要高带宽和低延迟,同时还可以容忍一定的过预订率。千兆网很适合。
- 计算网络 用于各计算节点的互连,是并行任务执行时的进程间通信的专用网络,并行计算的核心就是它和集群内其他节点交换信息的能力(IPC)。计算网络现在多用InfiniBand网络或者万兆以太网等。
- 存储网络 存储网络需要向HPC集群的节点提供数据访问服务。 有两种方法访问数据:一是数据由外部文件系统提供文件级别的访问,包括网络附属存储;二是数据块级别的访问,包括直连式存储或存储区域网络可以分别使用基于SCSI或SCSI RDMA协议的光纤通道或IB存储。
HPC的使用
登陆
登陆方式和普通的服务器基本一样,就是有一点:不同节点的IP地址不同。在Windows 的putty/Xshell或者Mac/Linux的terminal中登陆时注意使用的host name或IP要与节点node一致。例如此图中,login1和2是设定的登陆节点,如果用内网登陆login2,就用10.10.10.66IP;如果出差用外网,需要先配置VPN登陆到内网,然后再用192.168.1.24这个IP
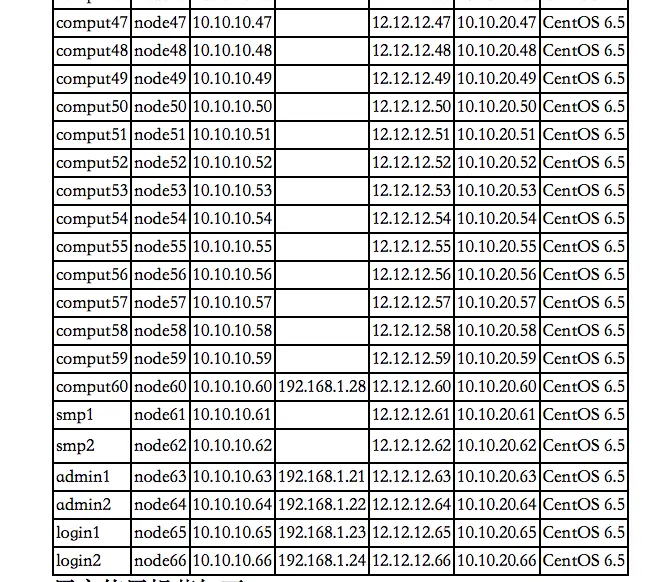
登陆以后,还是先修改密码,这里如果只输入passwd只是在当前节点修改了密码。如果想把所有的登陆节点有修改密码,需要先进入admin1节点ssh admin1 ,然后再passwd修改。
⚠️切记:不要在登陆节点处理大程序,一定要提交给计算节点;小的编译、测试程序可以
提交任务
提交任务的两大系统SGE与PBS【二者脚本相似,要清楚用的哪一个】
- SGE(Sun Grid Engine): 用户通过SGE索要计算资源,SGE相当于食堂打饭的工作人员,它本身并不生产资源,只是来分配。用户需要提供节点,也就是让分配管理者SGE知道你的打饭窗口
组成【了解即可】: 节点(Hosts/Nodes); 后台程序(Daemons) sge_qmaster – the Master Daemon sge_schedd – the Scheduler Daemon sge_execd – the Execution Daemon sge_commd – the Communicatoin Daemon 队列(Queues)
工作流程【了解】: a. 接受用户投放的任务; b. 任务运行以前,将任务放到一个存储区域; c. 发送任务到一个执行设备,并监控任务的运行; d. 运行结束写回结果并记录运行日志
投递任务脚本编写常用命令【重要】
模版:qsub [options] *.sh
[options]:
-cwd表示在当前路径下投放任务,任务的输出日志会输出到当前路径 【如果不设置,会在HOME下执行程序,并且结果都放在那里】-l vf=*G/M表示任务预估内存使用量,内存估计的值应稍微大于真实的内存-q指定要投放任务的队列;若不指定,SGE会选一个符合要求的-o -e分别制定标准输出和错误输出文件 【输出日志有两个:标准输出stfout 与 报错stderr】 【格式为:script.ojobID –> script是脚本名称,.o代表标准输出,jobID是作业ID号】-l h=node*指定使用哪个节点-pe smp *设定并行运行环境在节点内,任务占据的CPU个数 关于设定线程数: 有许多程序可以多线程运行,比如blastx --num_threads、GATK HaplotypeCaller -nct,假如需要20个线程,如果不通过qsub申请,就会占用所有的CPU资源,这样同一个节点下的用户就无法使用,严重时造成服务器宕机-hold_jid35,36,37意思是等待35-37号的任务结束后再开始执行-p设定优先级,优先级数字高的会让级别低的暂停,直接使用资源【一般需要管理员权限】-S指明命令解释器:SGE默认使用的是tcsh,而要投放的.sh是bash 【当然如果在qsub中不指定,也可以在脚本中第一行写#$ -S /bin/bash】查询任务
qstat -f查看所有任务qstat -s r仅检查正在执行的jobqstat -j jobID按任务号查看资源使用的退出情况qstat -u user按用户查看任务状态: qw 等待中 r 运行中 Eqw 投递任务出错 dr 节点挂掉了
删除任务
qdel -j XXX删除XXX号的任务bash脚本的魔数 魔数就是开头的注释信息,他让系统知道解释器的位置,投递任务前要加上
#! /bin/bash#$ -S /bin/bash
PBS(Portable Batch System) :它分配所需的硬件资源,用户不需要指定程序在哪写节点运行,系统根据资源空闲情况自动分配。比如还是去打饭,你只管坐下,说一句要吃什么,不需要自己去窗口,就可以等饭来。【例如曙光的Gridview就是PBS的修改版】
提交作业 使用pbs脚本,命令行直接qsub xxx.pbs
#一个pbs脚本实例,用vi编辑一个叫做bio.pbs的脚本或者叫配置文件 #每一行前面带有#PBS,从下一行开始: #PBS -N myjob #PBS -o /home/bio/mine.out #PBS -e /home/bio/mine.err #PBS -l nodes=2:ppn=2 cd 目录(程序所在的目录) *.sh-N name: 作业名称 -o path: 重定向标准输出到指定目录 -e path: 重定向错误输出到指定目录 -l resource_list: 制定资源列表,主要包括: nodes=N:ppn=M: 请求N个节点,每个节点M个处理器 mem=
N[K|M|G][B|W]:请求N {K|M|G}{bytes|words} 大小内存 -p priority: 任务优先级[-1024, 1023]。不定义为0 -r y|n : 指明作业是否可运行,y为可运行,n不可 配置完直接qsub bio.pbs就可以【如果想指定运行队列,使用qsub -q (队列名) *.pbs】
作业查询
qstat -a查看作业状态【会显示任务号jobID和任务进行状态】qstat -n查看作业使用节点qstat -f jobID查看具体信息作业删除
qdel + jobID