095-Bioconductor没想象的那么简单(part2)
刘小泽写于2019.3.24
今天看看Bioconductor的注释资源
前言
Bioconductor的注释信息有许多用处,比如可以将Ensembl ID转为HUGO symbol,但数据注释是一个复杂的工作,根据功能和位置主要有下图几种形式
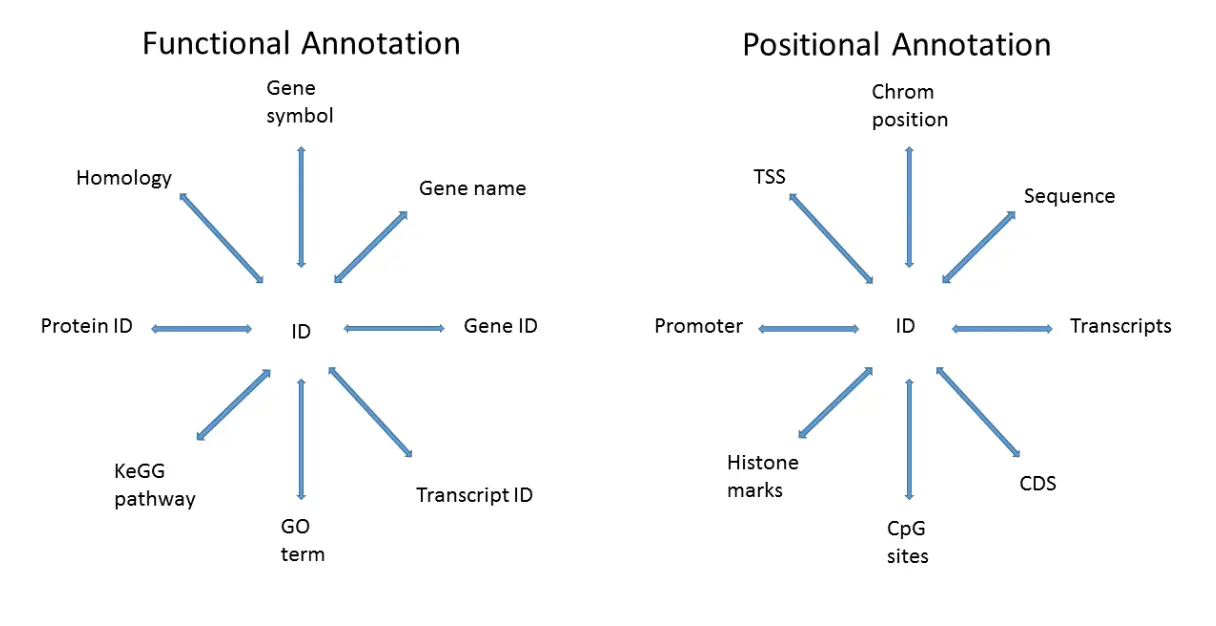
目前通常处理的生信数据格式主要有三类:实验数据(例如:表达矩阵Experimental data)、样本数据(Sample data)、注释数据(Annotation data)
可以存在三个不同的数据框,或者是ExpressionSet、SummarizedExperiment这样的对象
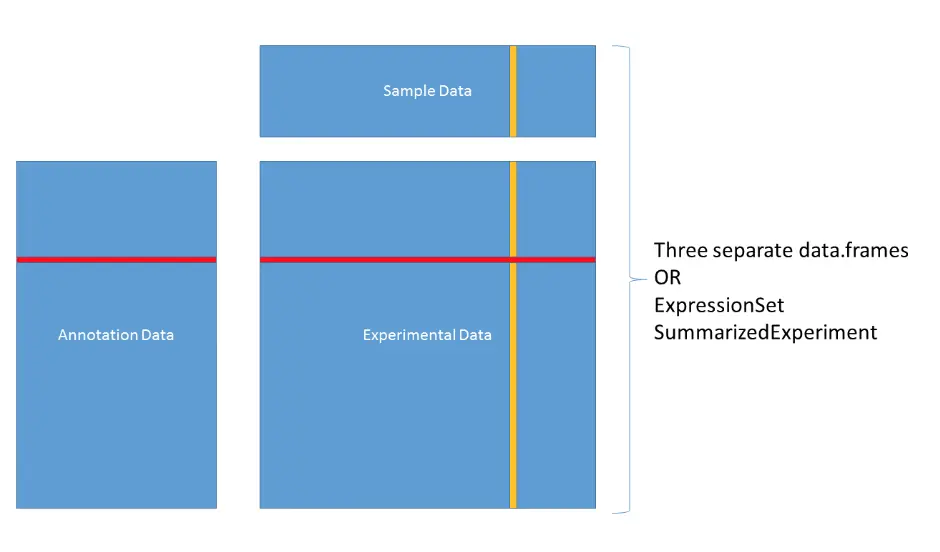
ExpressionSet
示例数据下载地址:https://github.com/jmacdon/Bioc2018Anno/blob/master/inst/extdata/eset.Rdata
> rm(list=ls())
> options(stringsAsFactors = F)
> load('eset.Rdata')
> eset
ExpressionSet (storageMode: lockedEnvironment)
assayData: 33552 features, 6 samples
element names: exprs
protocolData: none
phenoData
sampleNames: GSM2194079 GSM2194080 ... GSM2194084 (6 total)
varLabels: title characteristics_ch1.1
varMetadata: labelDescription
featureData
featureNames: 16657436 16657440 ... 17118478 (33552 total)
fvarLabels: PROBEID ENTREZID SYMBOL GENENAME
fvarMetadata: labelDescription
experimentData: use 'experimentData(object)'
Annotation: pd.hugene.2.0.st
eset中主要包含assayData、phenoData、featureData等
assayData: 表达矩阵(行为基因/探针名,列为样本),利用exprs提取 phenoData: 是AnnotatedDataFrame的一种,它的行数等于assayData的列数,相当于一些样本信息[上图中的Sample Data]; **featureData:**也是AnnotatedDataFrame的一种,它的行数等于assayData的行数,并且行名也相同,相当于一些基因信息[上图中的AnnotationData]
实验数据利用exprs取数据,利用pData看表型
> head(exprs(eset))
GSM2194079 GSM2194080 GSM2194081 GSM2194082 GSM2194083 GSM2194084
16657436 8.505158 9.046577 8.382674 9.115481 8.715343 8.566301
16657440 7.948860 8.191222 7.901911 8.459781 8.191793 8.219658
16657450 10.932934 11.228553 10.948120 11.462231 11.300046 11.300886
16657469 9.172462 9.344630 9.193450 9.465584 9.464020 9.135715
16657473 6.222049 6.551035 6.000246 6.398798 5.892654 5.592125
16657476 8.514300 8.474073 8.407196 8.811238 8.780833 8.874606
> head(pData(phenoData(eset)))
title characteristics_ch1.1
GSM2194079 SW620-miR625-rep1 shRNA: miR-625-3p
GSM2194080 SW620-miR625-rep2 shRNA: miR-625-3p
GSM2194081 SW620-miR625-rep3 shRNA: miR-625-3p
GSM2194082 SW620-scramble-rep1 shRNA: scramble
GSM2194083 SW620-scramble-rep2 shRNA: scramble
GSM2194084 SW620-scramble-rep3 shRNA: scramble
> head(pData(featureData(eset)))
PROBEID ENTREZID SYMBOL
16657436 16657436 84771 DDX11L2
16657440 16657440 100302278 MIR1302-2
16657450 16657450 402483 LINC01000
16657469 16657469 140849 LINC00266-1
16657473 16657473 729759 OR4F29
16657476 16657476 388574 RPL23AP87
GENENAME
16657436 DEAD/H-box helicase 11 like 2
16657440 microRNA 1302-2
16657450 long intergenic non-protein coding RNA 1000
16657469 long intergenic non-protein coding RNA 266-1
16657473 olfactory receptor family 4 subfamily F member 29
16657476 ribosomal protein L23a pseudogene 87
注释资源

使用AnnoDb包
主要函数就是select => select(annopkg, keys, columns, keytype)
其中:annopkg是注释包;keys是目前知道的ID号;columns是想得到的列;
keytype是使用的key的类型
简答的例子
分析Affymetrix Human Gene ST 2.0 array芯片数据,然后想知道这些基因是什么
> BiocManager::install("hugene20sttranscriptcluster.db", version = "3.8") > library(hugene20sttranscriptcluster.db) > set.seed(12345) # 随机选择一些probe ID > ids <- featureNames(eset)[sample(1:25000, 5)] > ids [1] "16715361" "16774497" "16836414" "16910671" "17002888" # 看看probe ID对应的基因名 > AnnotationDbi::select(hugene20sttranscriptcluster.db, ids, "SYMBOL") 'select()' returned 1:1 mapping between keys and columns PROBEID SYMBOL 1 16908472 LINC01494 2 16962185 ALG3 3 16920686 <NA> 4 16965513 <NA> 5 16819952 CBFB我们怎么知道注释包中的核心ID(central keys)呢?比如在上面的芯片注释包(ChipDb)中,核心ID就是探针ID(probe ID); 如果是物种注释包(例如org.Hs.eg.db),核心ID就是
eg代表的Entrez Gene ID;即使不知道核心ID是什么类型也没关系,使用
keytypes()或columns()可以看到包中所有的ID类型,然后利用select选出这些ID> if (! require(hgu95av2.db,character.only=T) ) { BiocManager::install(hgu95av2.db,ask = F,update = F) } > columns(hgu95av2.db) # or keytypes(hgu95av2.db) > keys <- head( keys(hgu95av2.db) ) > keys > AnnotationDbi::select(hgu95av2.db, keys=keys, columns = c("SYMBOL","ENTREZID")) 'select()' returned 1:1 mapping between keys and columns PROBEID SYMBOL ENTREZID 1 1000_at MAPK3 5595 2 1001_at TIE1 7075 3 1002_f_at CYP2C19 1557 4 1003_s_at CXCR5 643 5 1004_at CXCR5 643 6 1005_at DUSP1 1843再看一个例子
> AnnotationDbi::select(hugene20sttranscriptcluster.db, ids, c("SYMBOL","MAP")) 'select()' returned 1:many mapping between keys and columns PROBEID SYMBOL MAP 1 16737401 TRAF6 11p12 2 16657436 DDX11L1 1p36.33 3 16657436 LOC102725121 1p36.33 4 16657436 DDX11L2 2q14.1 5 16657436 DDX11L9 15q26.3 6 16657436 DDX11L10 16p13.3 7 16657436 DDX11L5 9p24.3 8 16657436 DDX11L16 Xq28 9 16657436 DDX11L16 Yq12 10 16678303 ARF1 1q42.13select的替换=》mapIDs从上面的例子中可以看到,
select的结果存在一些重复,利用mapIDs可以进行控制。但两者的参数还有略微不同:> select(x, keys, columns, keytype, ...) > mapIds(x, keys, column, keytype, ..., multiVals) # 解释columns:the columns or kinds of things that can be retrieved from the database # 解释keytype:the keytype that matches the keys used.mapIDs只能指定一个column;必须指定keytype;最后一个multiVals参数是为了控制重复> mapIds(hugene20sttranscriptcluster.db, ids, "SYMBOL", "PROBEID") 'select()' returned 1:many mapping between keys and columns 16737401 16657436 16678303 "TRAF6" "DDX11L1" "ARF1"mapIDs中multiVals参数的设定默认使用
multiVals = first,表示遇到重复只取第一个;另外还有list、CharacterList、filter、asNA等> mapIds(hugene20sttranscriptcluster.db, ids, "SYMBOL", "PROBEID",multiVals = "list") 'select()' returned 1:many mapping between keys and columns $`16737401` [1] "TRAF6" $`16657436` [1] "DDX11L1" "LOC102725121" "DDX11L2" "DDX11L9" [5] "DDX11L10" "DDX11L5" "DDX11L16" $`16678303` [1] "ARF1" > mapIds(hugene20sttranscriptcluster.db, ids, "SYMBOL", "PROBEID",multiVals = "CharacterList") 'select()' returned 1:many mapping between keys and columns CharacterList of length 3 [["16737401"]] TRAF6 [["16657436"]] DDX11L1 LOC102725121 DDX11L2 DDX11L9 DDX11L10 DDX11L5 DDX11L16 [["16678303"]] ARF1 > mapIds(hugene20sttranscriptcluster.db, ids, "SYMBOL", "PROBEID",multiVals = "filter") # 只返回没有重复的结果 'select()' returned 1:many mapping between keys and columns 16737401 16678303 "TRAF6" "ARF1" > mapIds(hugene20sttranscriptcluster.db, ids, "SYMBOL", "PROBEID",multiVals = "asNA") #把重复结果标记为NA 'select()' returned 1:many mapping between keys and columns 16737401 16657436 16678303 "TRAF6" NA "ARF1"芯片/物种数据库的小练习
芯片注释库以
hugene20sttranscriptcluster.db为例;物种注释库以org.Hs.eg.db为例,下面以物种注释库为例:# What gene symbol corresponds to Entrez Gene ID 1000? > mapIds(org.Hs.eg.db, "1000", "SYMBOL", "ENTREZID") 'select()' returned 1:1 mapping between keys and columns 1000 "CDH2" # What is the Ensembl Gene ID for PPARG? > mapIds(org.Hs.eg.db, "PPARG", "ENTREZID", "SYMBOL") 'select()' returned 1:1 mapping between keys and columns PPARG "5468" # What is the UniProt ID for GAPDH? > mapIds(org.Hs.eg.db, "GAPDH", "UNIPROT", "SYMBOL") 'select()' returned 1:many mapping between keys and columns GAPDH "P04406"最后,看一下之前的探针数据
eset有多少探针只比对到一个基因id?又有多少没有比对上基因id?致谢: 花花有句名言:“所有能放在数据框的东西,都难不倒我,不是吹牛!" 于是下面的dplyr的方法就是花花支招
> mpid <- mapIds(hugene20sttranscriptcluster.db, rownames(eset), "ENTREZID", "PROBEID") > length(mpid) [1] 33552 # 只出现一次[方法一:dplyr] > count(tmp2,mpid,sort = T) %>% filter(n==1) %>% nrow() [1] 22586 # 只出现一次[方法二:base] >sum(table(mpid)==1) [1] 22586 # Don’t map to a gene at all(也就是统计NA值)[方法一:base] > tmp <- as.data.frame(mpid) > sum(is.na(tmp$mpid)) [1] 7900 # 另一种统计NA方法[方法二:dplyr] > count(tmp2,mpid) # 可以从tibble中直接看到NA数量 # A tibble: 23,573 x 2 mpid n <chr> <int> 1 NA 7900这里我也感悟到,使用tidy data进行数据处理,前期可能比较麻烦,因为需要把数据转成要求的格式,但是后来的结果会比较好处理
TxDb包:包含了位置信息,并且其中的内容可以通过包的名字推断出来例如:
TxDb.Hsapiens.UCSC.hg19.knownGene其中的字段包含了:人类;UCSC;hg19(GRCh37);已知基因
类似的还有:
TxDb.Dmelanogaster.UCSC.dm3.ensGene;TxDb.Athaliana.BioMart.plantsmart22> AnnotationDbi::select(TxDb.Hsapiens.UCSC.hg19.knownGene, c("1","10"), + c("TXNAME","TXCHROM","TXSTART","TXEND"), "GENEID") 'select()' returned 1:many mapping between keys and columns GENEID TXNAME TXCHROM TXSTART TXEND 1 1 uc002qsd.4 chr19 58858172 58864865 2 1 uc002qsf.2 chr19 58859832 58874214 3 10 uc003wyw.1 chr8 18248755 18258723EnsDb包:与TxDb包相似,但基于Ensembl比对,命名形式比如:EnsDb.Hsapiens.v79;EnsDb.Mmusculus.v79;EnsDb.Rnorvegicus.v79> AnnotationDbi::select(EnsDb.Hsapiens.v86, c("1", "10"), + c("GENEID","GENENAME","SEQNAME","GENESEQSTART","GENESEQEND"), "ENTREZID") ENTREZID GENEID GENENAME SEQNAME GENESEQSTART GENESEQEND 1 1 ENSG00000121410 A1BG 19 58345178 58353499 2 10 ENSG00000156006 NAT2 8 18391245 18401218