131-简述Bioconductor的注释包AnnotationDbi
刘小泽写于19.8.5 AnnotationDbi应该是进行富集分析时最常用的注释包了,所以有必要好好了解一下
简介
这是我们经常用到的注释库,看似是一个包,实则是一个数据库;这里面的包加载进来就是一个Bimap对象,它可以将一组keys关联到另一组keys,起到一个桥梁的作用。这个对象以后使用toTable函数转换成数据框
对于这个Bimap对象,还需要多说一句
# 利用get直接获取某个关联结果;
> get("1553505_at",hgu133plus2SYMBOL)
[1] "A2ML1"
# mget就是multi-get,获取多个关联结果
> mget(c("1553545_at","1553505_at"),hgu133plus2SYMBOL,ifnotfound = NA)
$`1553545_at`
[1] "ILDR1"
$`1553505_at`
[1] "A2ML1"
包含内容
基于物种
例如
org.Hs.eg.db,像这种物种数据库一共有19个物种(https://bioconductor.org/packages/release/BiocViews.html#___OrgDb)使用
org.Hs.eg_dbInfo()可以看到这个人类的注释数据库的信息: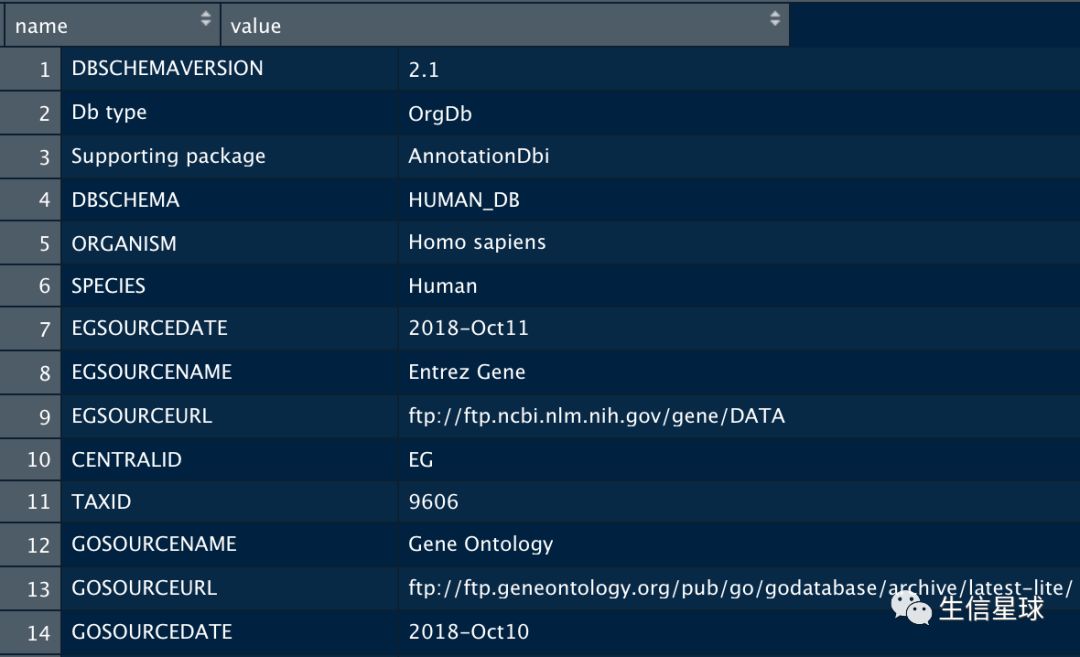
看包含什么内容
> columns(org.Hs.eg.db) [1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT" [5] "ENSEMBLTRANS" "ENTREZID" "ENZYME" "EVIDENCE" [9] "EVIDENCEALL" "GENENAME" "GO" "GOALL" [13] "IPI" "MAP" "OMIM" "ONTOLOGY" [17] "ONTOLOGYALL" "PATH" "PFAM" "PMID" [21] "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG" [25] "UNIGENE" "UNIPROT"其中,PATH就是KEGG pathway IDs,ALIAS就是基因别名,其他的顾名思义
如果要选择其中某一部分(利用
toTable或者select)来自
select的帮助文档:select,columnsandkeysare used together to extract data from anAnnotationDbobject# 方法一 head(toTable(org.Hs.egSYMBOL2EG),6) # 方法二 keys <- head( keys(org.Hs.eg.db) ) select(org.Hs.eg.db, keys=keys, columns = c("SYMBOL"))
基于平台
例如hgu133plus2.db ,这一类在芯片数据中应用较多,例如hgu95av2.db是hgu95av2 Affymetrix 的芯片
# 查看详细信息
hgu133plus2_dbInfo()
# 也是支持keys(), columns() and select()
基于系统框架
例如GO.db、KEGG.db
小练习
安装hgu95av2包并找到1553545_at探针的 gene symbol, chromosome position and KEGG pathway ID
# 提示:hgu95av2CHRLOC、hgu95av2PATH
get("1003_s_at",hgu95av2SYMBOL)
大练习
以人类为例,探索GO term与gene id的互换关系,将全部以R代码展示
library(org.Hs.eg.db)
# 官方教程:https://bioconductor.org/packages/release/data/annotation/manuals/org.Hs.eg.db/man/org.Hs.eg.db.pdf
# 我们接下来使用 org.Hs.egGO (它是entrez ID和GO ID的对应)
entrez_object <- org.Hs.egGO
> class(entrez_object)
[1] "Go3AnnDbBimap"
attr(,"package")
[1] "AnnotationDbi"
# 在org.Hs.egGO其中找到具有GO ID的Entrez ID
mapped_genes <- mappedkeys(entrez_object)
> head(mapped_genes)
[1] "1" "10" "100" "1000" "10000" "10001"
# 将每个gene ID对应的GO ID分别提取对应到gene ID上,并存为列表(因为长度不一)
entrez_to_go <- as.list(entrez_object[mapped_genes])
> length(entrez_to_go)
[1] 20408
# 举个例子:以Entrez ID=1为例(https://www.ncbi.nlm.nih.gov/gene/?term=1),它的Gene Symbol是A1BG。以下列出的就是这个基因相关的GO terms
> sapply(entrez_to_go[[1]], function(x) x)
GO:0002576 GO:0008150 GO:0043312 GO:0005576
GOID "GO:0002576" "GO:0008150" "GO:0043312" "GO:0005576"
Evidence "TAS" "ND" "TAS" "HDA"
Ontology "BP" "BP" "BP" "CC"
GO:0005576 GO:0005576 GO:0005615 GO:0031093
GOID "GO:0005576" "GO:0005576" "GO:0005615" "GO:0031093"
Evidence "IDA" "TAS" "HDA" "TAS"
Ontology "CC" "CC" "CC" "CC"
GO:0034774 GO:0062023 GO:0070062 GO:0072562
GOID "GO:0034774" "GO:0062023" "GO:0070062" "GO:0072562"
Evidence "TAS" "HDA" "HDA" "HDA"
Ontology "CC" "CC" "CC" "CC"
GO:1904813 GO:0003674
GOID "GO:1904813" "GO:0003674"
Evidence "TAS" "ND"
Ontology "CC" "MF"
# 当然,如果反过来,想按照GO ID 找对应的gene ID也是可以的
go_object <- as.list(org.Hs.egGO2EG)
> length(go_object)
[1] 17983
# 其中第一个就是GO:0000002,它对应的意思是:mitochondrial genome maintenance
> go_object[1]
$`GO:0000002`
TAS IMP ISS IMP IMP NAS IMP IBA IDA
"291" "1890" "4205" "4358" "4976" "9361" "10000" "55154" "55186"
IEA IDA IMP
"80119" "84275" "92667"
# 这个结果要注意一下:结果引号中的是基因ID,例如291就是SLC25A4基因(https://www.ncbi.nlm.nih.gov/gene/?term=291)。然后TAS、IMP、ISS等等是GO evidence codes(参考:http://geneontology.org/docs/guide-go-evidence-codes/)
# 好,现在找出GO:0007411 (axon guidance)的全部基因
axon_gene <- go_object['GO:0007411']
> length(unlist(axon_gene, use.names=FALSE))
[1] 201
# 但并非所有基因都只出现了一次(查看只出现一次的基因用unique函数)
> length(unique(unlist(axon_gene, use.names=FALSE)))
[1] 191
# 最后只用unique gene
> axon_gene <- unique(unlist(axon_gene, use.names=FALSE))
> head(axon_gene)
[1] "323" "474" "627" "655" "682" "1002"