120-Bioconductor没想象的那么简单-part9
刘小泽写于19.5.28 跟着Bioconductor书再看一遍他们是如何介绍GenomicRanges的,不同的作者不同的见解,也有不同的教学方法。多学多看,这一部分内容来自 Bioconductor 2018的第四章 part1:https://www.jianshu.com/p/b030d05a80a4 part2:https://www.jianshu.com/p/c4fdab9929db part3:https://www.jianshu.com/p/f43d9d07a4af part4:https://www.jianshu.com/p/4560a29d8da2 part5:https://www.jianshu.com/p/e695b4a1dcf7 part6:https://www.jianshu.com/p/79ad7355fb8c part7:https://www.jianshu.com/p/37448531b533 part8:https://www.jianshu.com/p/8f7f20371116
前言
什么是Ranges基础?
它就是处理组学数据使用的,包含了数据结构和算法。它包含了标准的基因组数据"容器“(像GRanges和SummarizedExperiment),优化的数据重现方式(Rle),还有快速的算法(像计算overlap、找近邻位点、统计区间信息)。可以看到,这是一个很大的整体,其中我们 之前接触到的GRanges只是其中一小部分。
为什么要学Ranges基础?
因为数百个R包都是建立在Ranges基础上的,它可以方便整合多个包、多种数据类型以构建一个流程。代码拓展性强(可以从流程脚本向R包脚本转换)。
入门——GenomicRanges
核心的数据结构就是GenomicRanges,它包含了许多代表基因组位置的信息,一般是起始和终止信息(当然这个位置信息的重合是少不了的,这会给统计一些基础信息带来困扰,这个问题之后再说)。除了这个,还可以包含结合位点、序列比对、转录本信息等。
实战
生成
最简单的生成GRanges方法应该就是数据框转换了
library(GenomicRanges)
df <- data.frame(
seqnames = rep(c("chr1", "chr2", "chr1", "chr3"), c(1, 3, 2, 4)), start = c(101, 105, 125, 132, 134, 152, 153, 160, 166, 170),
end = c(104, 120, 133, 132, 155, 154, 159, 166, 171, 190),
strand = rep(strand(c("-", "+", "*", "+", "-")), c(1, 2, 2, 3, 2)), score = 1:10,
GC = seq(1, 0, length=10),
row.names = head(letters, 10))
View(df)
gr <- makeGRangesFromDataFrame(df, keep.extra.columns=TRUE)
这样的结果就是建立了包含10个ranges的GRanges对象
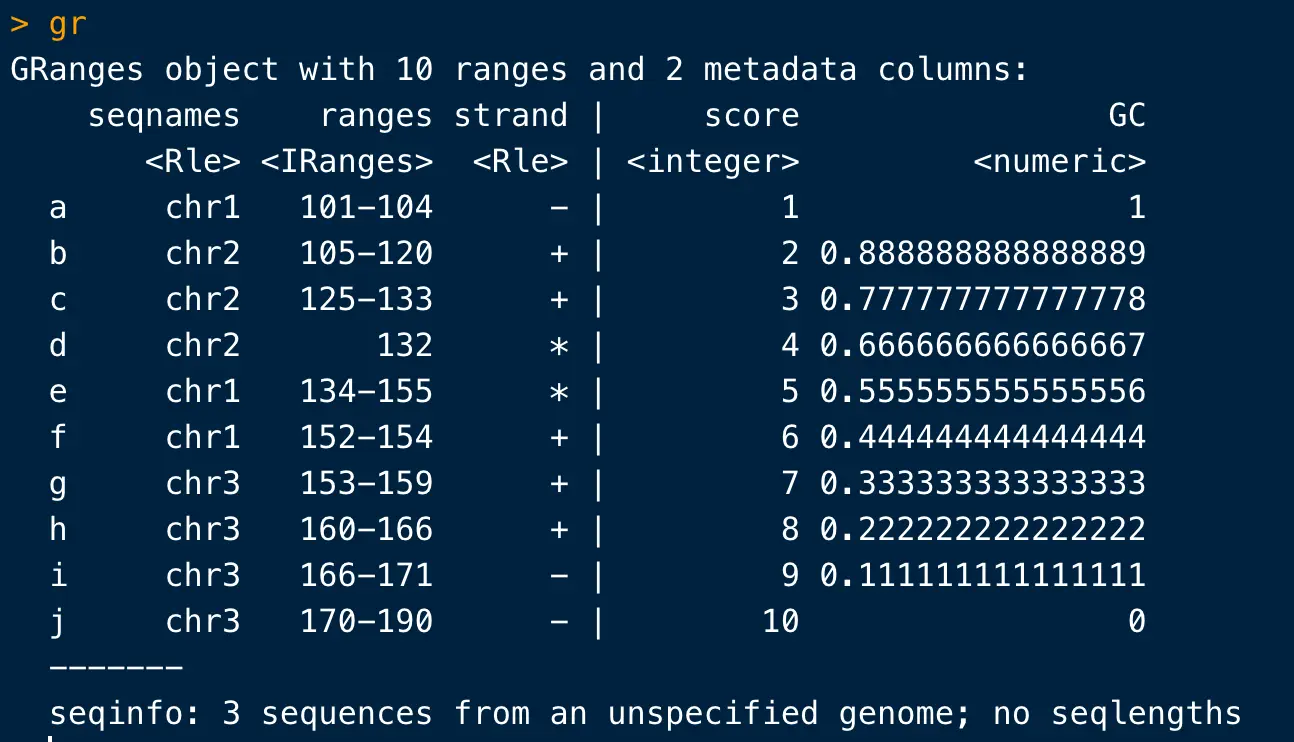
之前介绍过这里,以|为分隔,左边是基因组坐标信息(序列名、范围、链),右边是其他的注释信息(这里是score和GC含量,当然它可以容纳更多类型的数据)
也可以从标准文件格式(如:BED、GFF、BigWig)导入为GRanges,利用
rtracklayer包,part7:https://www.jianshu.com/p/37448531b533有过介绍另外,利用
GenomicAlignments包可以利用BAM文件构建GAlignments对象,这个对象和GRanges很像,当然也可以转换成GRanges
查看
对左侧基本信息操作
# 先看三大项(名称、区间、链)
seqnames(gr)
ranges(gr)
strand(gr)
# 另外,可以同时看这三项,并排除其余的metadata(也就是只返回竖线左侧的内容)
granges(gr)
# 如果要分别查看起始、终止、区间位置(也就是把上面ranges函数的结果拆开)
start(gr)
end(gr)
width(gr)
对右侧注释信息操作
mcols(gr) #将metadata返回为数据框,当然可以直接从结果中选取某一列
mcols(gr)$score
#上面结果等同于 score(gr)
总体操作
# 看行名
names(gr)
# 看多少行
length(gr)
取子集
这个对象取子集的方法和向量的操作类似,可以直接gr[2:3]
取出来是这样的:

然后我们可以指定后面取出哪个metadata:
> gr[2:3, "GC"]
GRanges object with 2 ranges and 1 metadata column:
seqnames ranges strand | GC
<Rle> <IRanges> <Rle> | <numeric>
b chr2 105-120 + | 0.888888888888889
c chr2 125-133 + | 0.777777777777778
当然这只是一种最基础的办法,更方便的办法如:subset()
> subset(gr, strand == "+" & score > 5, select = score)
GRanges object with 3 ranges and 1 metadata column:
seqnames ranges strand | score
<Rle> <IRanges> <Rle> | <integer>
f chr1 152-154 + | 6
g chr3 153-159 + | 7
h chr3 160-166 + | 8
对GRanges对象的修改
比如可以复制并替换一行
grMod <- gr
grMod[2] <- gr[1]
# 结果新的grMod对象的第二行就和第一行一样
还可以对行进行重复、反转、选特定行
rep(gr[2], times = 3) # 重复第二行3次
rev(gr) #将原来的所有行反转
head(gr,n=2) # 从头取
tail(gr,n=2) # 从尾端取
window(gr, start=2,end=4) # 从中间连续取
gr[IRanges(start=c(2,7),end=c(3,9))] # 从中间不连续取(也就是取出2~3行 + 7~9行)
拆分与合并
拆分用split(),结果返回一个列表GRangesList 【很好记:对象下面是列表,列表下面是数据框,数据框下面是向量】。只不过这里的列表中包含的还是小对象(而不是一般意义上的数据框)
首先最直观的是按行拆分
sp <- split(gr, rep(1:2, each=5))
sp
# 这个意思就是说:将原来的对象按rep(1:2, each=5)这个格式拆,而这个格式是1 1 1 1 1 2 2 2 2 2一共10个数,因此这里也就明白,只要我们告诉split函数我们要拆分的结果是怎么组合的,就可以想怎么拆就怎么拆
另外除了按行,还可以按注释信息metadata拆分
split(gr, ~ strand)
# 这个结果就分成了正链、负链和未知链三个
合并用c()、append()
# 因为之前拆分的结果是一个列表,那么选择其中的元素还是要用[[]]
c(sp[[1]], sp[[2]])
另外stack()函数可以将GRangesList 列表中的小对象重新组装到一起,并可以为每个小对象添加一个分组信息
stack(sp, index.var="group")
整合统计
利用aggregate函数,其中要指定一个公式y ~ x (x表示按什么分组,y表示分组后每组的值)
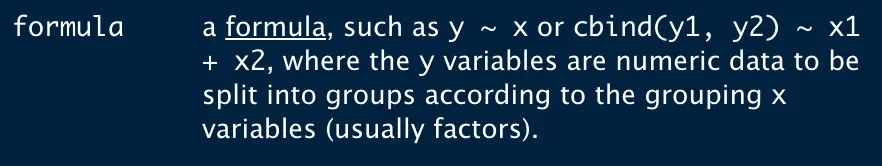
有两种方法进行统计:
# 第一种
aggregate(gr, score ~ strand, mean)
# 第二种
aggregate(gr, ~ strand, n_score = lengths(score), mean_score = mean(score))
# 注意到其中统计长度用的是lengths而不是length,因为这里是先按照strand进行分组,然后再统计score长度与均值信息。因此score的信息是类似于一个列表,其中有正链的score信息、负链的score信息以及未知链的score信息,如果我们对其中任一个score信息进行长度统计,是可以用length()的,但是放在一起统计,就要用lengths()