121-不要"太重视“fastqc的结果
刘小泽写于19.5.29 原始数据质控永远是好的分析结果的第一步,我们主流都是采用fastqc软件,结果会产生一个html网页,下载下来看看就知道大体的数据质量。但是,其中这么多内容,我都应该相信吗? 答案是NO!因此这里题目用引号也表示强调,不要因为看到summary中出现了几个叉号就对自己的数据不报希望,或者直接无视所有的结果,这样都是不对的,还是要了解自己的数据
1 使用fastqc质控的意义
首先,要知道目前主要使用的二代测序仪主要是Illumina出品,它的特点有两个:桥式扩增和边合成边测序(sequencing-by-synthesis,SBS)
它的第二个特点主要依靠化学反应来完成碱基合成的过程,使得从5‘=》3’可以不断合成延伸。那么既然是化学反应,就一定存在扩增效率减弱的现象(因为聚类酶的活性在降低,导致结合的特异性变差),因此测序长度越长,后面数据的质量会降低的越厉害
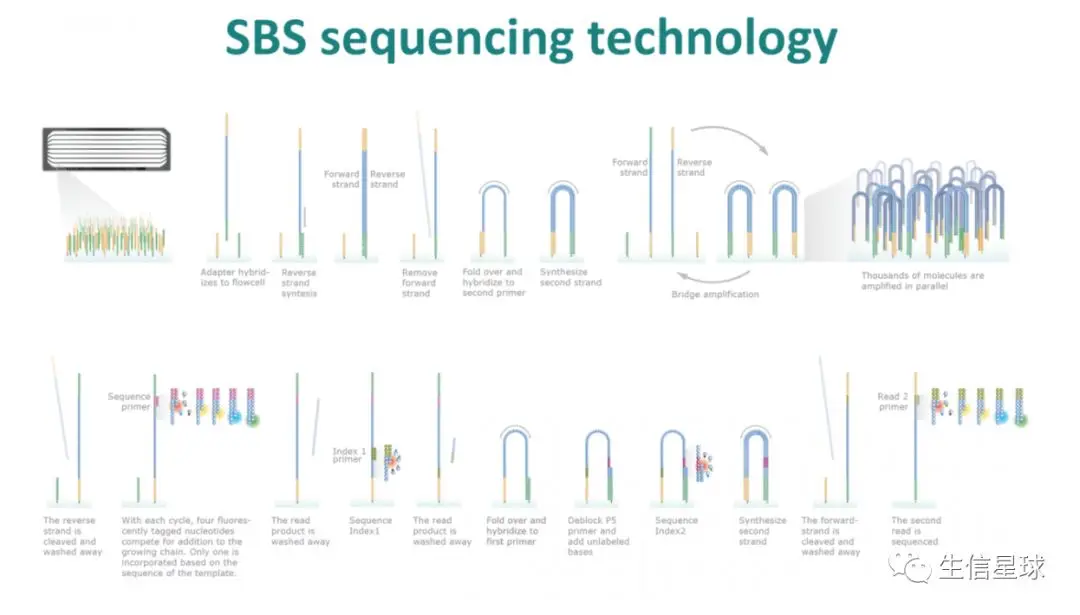
(图片来源:http://www.biofidal-lab.com/details-miseq+illumina+sequencing+system+at+lyon-50.html)
我们知道fastq文件的格式,其中第四列表示碱基质量值,如果要对一条reads求它的质量值(也就是Phred值),可以利用python的ord()函数(https://www.biostars.org/p/225923/)
这个ord函数将字符转为ASCII对应的数字,然后目前我们大多使用的测序仪都是Phred33标准,再减去这个33,就得到了最后的质量值
python -c 'from math import*; print 10**-((ord("A")-33)/10.0)'
但是,这仅仅是对一条read求解,但是一般一个样本的测序结果就要十几M甚至几十M,也就是几千万条reads,即使用批量处理也很慢,因此fastqc就专门解决这个问题,最后以可视化形式来展现,而且它展示的是全部reads的共同结果,而不是某一条read,方便全局把握数据质量
2 fastqc的结果很多,该看什么?
官网已经很详细的介绍了每部分的含义:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/
这里只是列出一些重点,防止眼花缭乱找不到重点
其实一开始接触这个软件的时候,我还记得当时我是把所有的图都看了一遍,结果除了第一个summary和总体质量值的那张图外,其余都看不懂
好,话不多说,从fastqc结果的第一部分开始看:
首先映入眼帘的是summary,也就是这个软件对你的数据做的所有的事(也就是所有的模块——module)
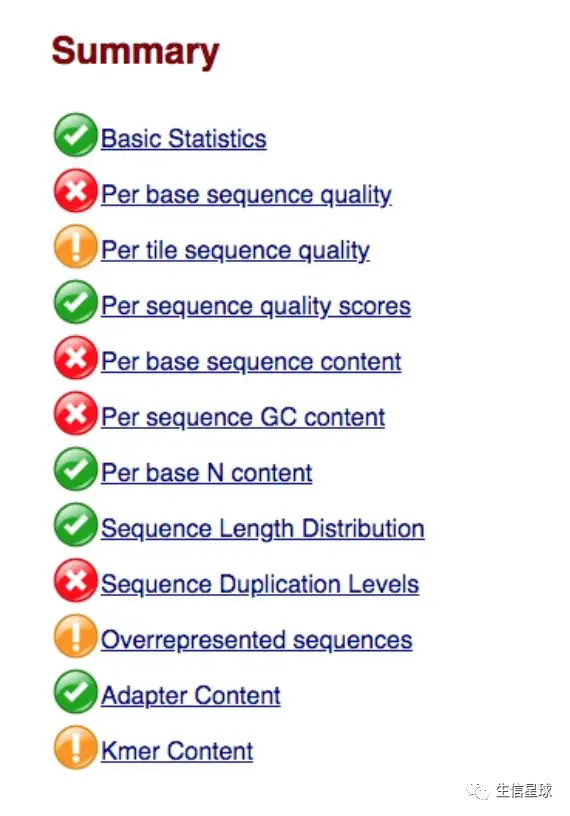
其中黄色表示Warning,红色表示Fail
不得不说,软件这里做的可能容易产生误导,因为它认为与它的标准相差太大就给红牌罚下,这样对所有的数据"一视同仁"有些武断,最好应该像"挖地雷"游戏中,标记成小旗,让我们知道这里有待商榷就好
2.1 第一个模块 Basic Statistics
样本的基础信息,有用的是箭头标出的部分

2.2 第二个模块 Per base sequence quality
这是最重要的质控模块之一,它描述的是: Distribution of quality scores at each position in the read across all reads
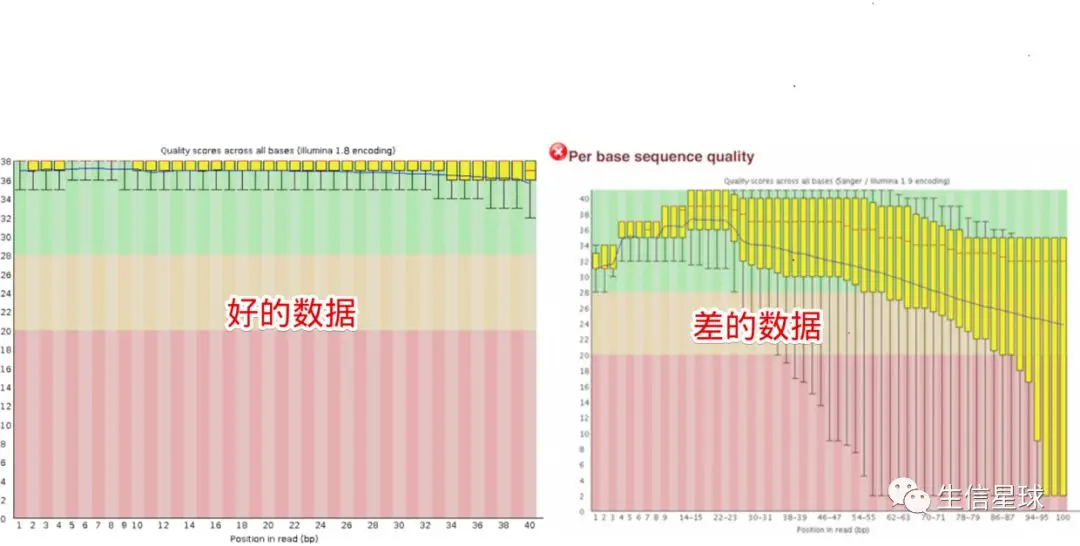
其中纵轴表示Phred碱基质量,横轴表示碱基在reads上的位置。比如:横坐标的1表示所有reads的第一个碱基的质量值分布,结果用一个箱线图表示。(如果不知道箱线图是什么?搜索一下,比如:https://whatis.techtarget.com/definition/box-plot)。箱线图的红线表示中位数,其余的四条线为10、25、75、90四分位数。
然后又看到有一条贯穿箱线图的蓝色线(看右图差的数据),它表示平均碱基质量值。
整个图又有三块背景:红色背景区域表示reads质量很差(Phred < 20),绿色背景表示质量不错(Phred > 28),因此看到右图中第一个碱基的箱线图中的10分位数也在纵轴的28以上,表示几乎全部的reads的第一个碱基质量值都大于28
不是所有的数据都那么幸运,会出来左边的结果。怕就怕,看到结果差,却不知道为什么差,更不用说从何下手去纠正这个问题了。 因此这里我们不看好的数据,只看差的数据的产生原因是什么
2.2.1 测序错误概览
对于Illumina测序来说,**碱基测序质量和荧光信号的强度与纯度有关(**signal intensity and purity of the fluorescent signal),因为它后续是用一个非常灵敏的照相机进行拍照。如果检测到的荧光强度太低或者一次检测出现了多种荧光信号,就会导致最后系统判断这个碱基质量值低。
测序错误又分为能预料到的和不能预料的:
能预料的错误:也就是我们知道测序仪从第一次扩增cycle到最后一个cycle一定存在下降的趋势,一般导致的因素有:信号减弱(Signal decay)、相位(Phasing)
信号减弱:由于荧光团的降解(之前DNA聚合时每个碱基都是带着荧光基团的,荧光降解当然会识别不出这个碱基);或者一个簇(cluster)中一定比例的reads没有延长成功(因此越往后,合成放出荧光的"力气"越来越小)
相位:原来为了保证一次只添加一个碱基,会在聚合的碱基中提前加上3’ terminators和荧光基团。结合并检测到荧光后,再将这个碱基去掉,进行下一个。但是如果说3’ terminators和荧光基团没有成功去除或者说加的3’ terminators根本不好用,那么这个碱基就不会被去掉,它会一直向下聚合并发出和原来一样的荧光
具体原理可以看:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2745764/
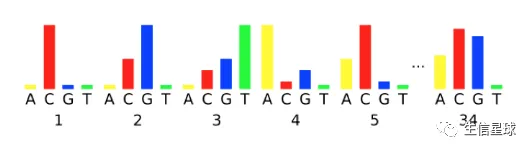
可以看到,越到后来的循环,越不容易看到真实的荧光
不能预料的错误: 簇的交叉(Overclustering)和仪器运行错误(Instrumentation breakdown)
- 簇的交叉:当每个扩增簇离得很近,它们可能会通过桥式扩增产生交叉,这原本的两个簇最后会被当成一个簇来解读荧光信号,降低了荧光强度,将整个read质量值降低
- 仪器问题:比如说reads质量值本来非常好,然后质量值突然呈90度下降;再比如绝大部分都是低质量reads
有个网站记录了许多qc结果差的例子:http://bioinfo-core.org/index.php/9th_Discussion-28_October_2010
2.2.2 第一种坏数据

过去测得越长,越容易出现碱基质量逐步下降这个问题。新的试剂研发(比如聚合酶稳定性更好)缓解了一下这种问题,但同时需求的碱基长度也越来越长。一般认为,质量值中位数降到20以下是要考虑剪切(trim)掉的,因为特别低质量的reads在后续分析中不好修复,反而会引入更大误差
2.2.3 第二种坏数据
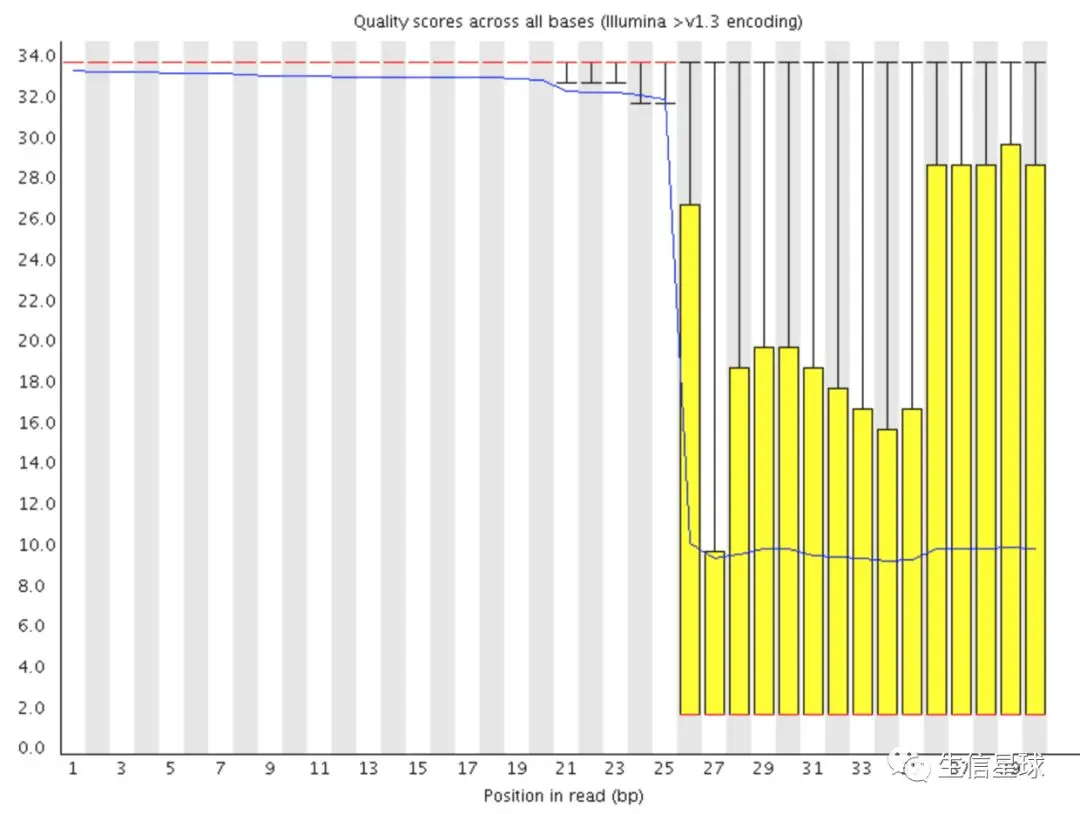
像这种突然从好数据急转到坏数据的情况,很有可能是测序仪的问题
那么好,既然我们看了上面👆数据存在问题的几种原因,再回过头来,看看之前右边的这个差的数据
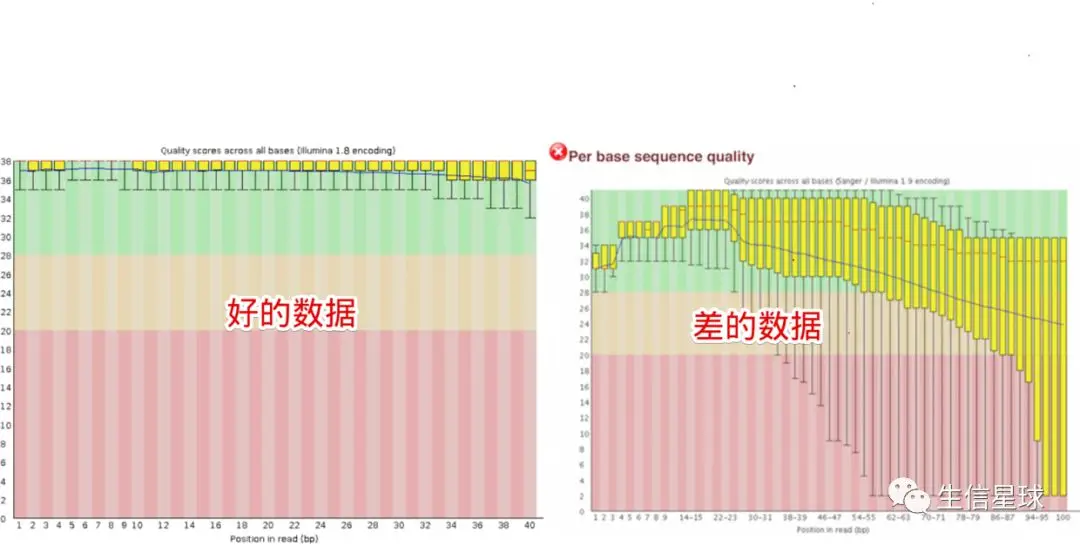
可以看到,质量值是逐渐下降的,到尾部降到最低,可以解释为信号减弱或者相位产生;而由于机器问题可能性不大,因此去掉低质量的reads后还是可以用的
2.3 碱基总体质量值分布 Per sequence quality scores
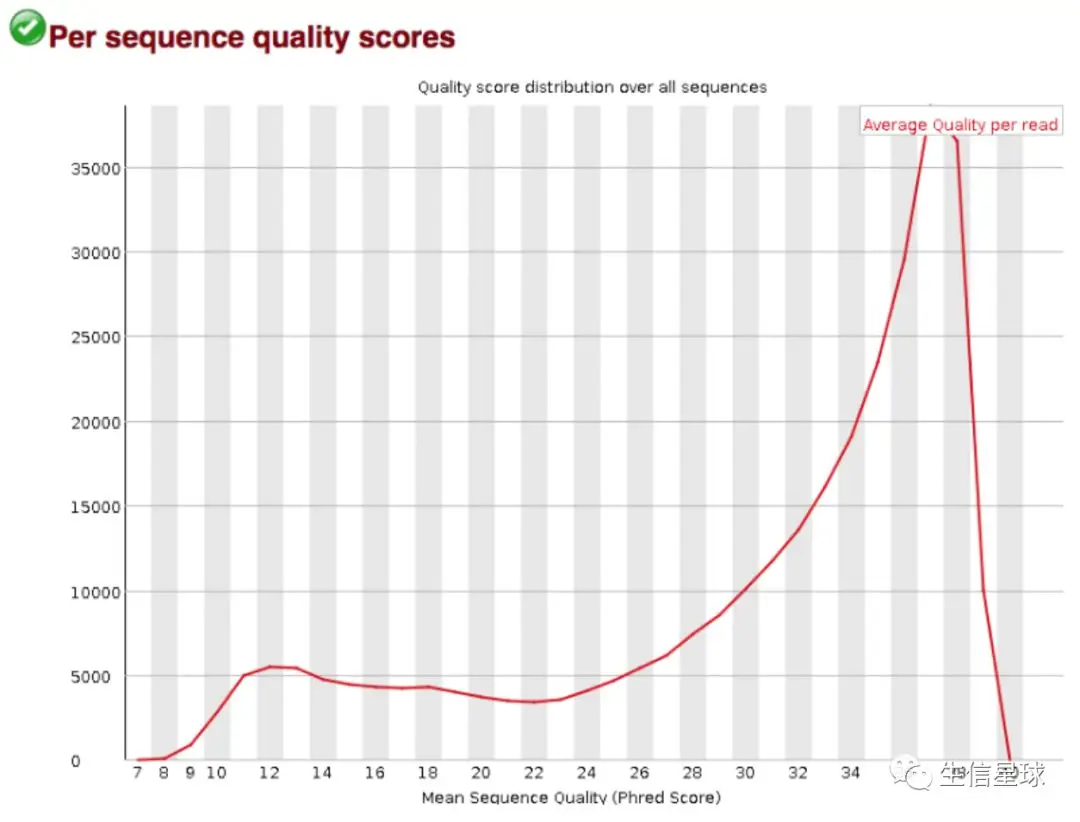
它的横轴是平均质量分数,纵轴是测序reads数。我们比较希望在低质量区域(也就是x轴前端)没有较大的峰(也就是没有太多的reads)
这个图中可以看到:在质量值为12时有一个小突起,但数量不是特别多,因此问题不大
2.4 read各个位置上碱基比例分布 Per base sequence content
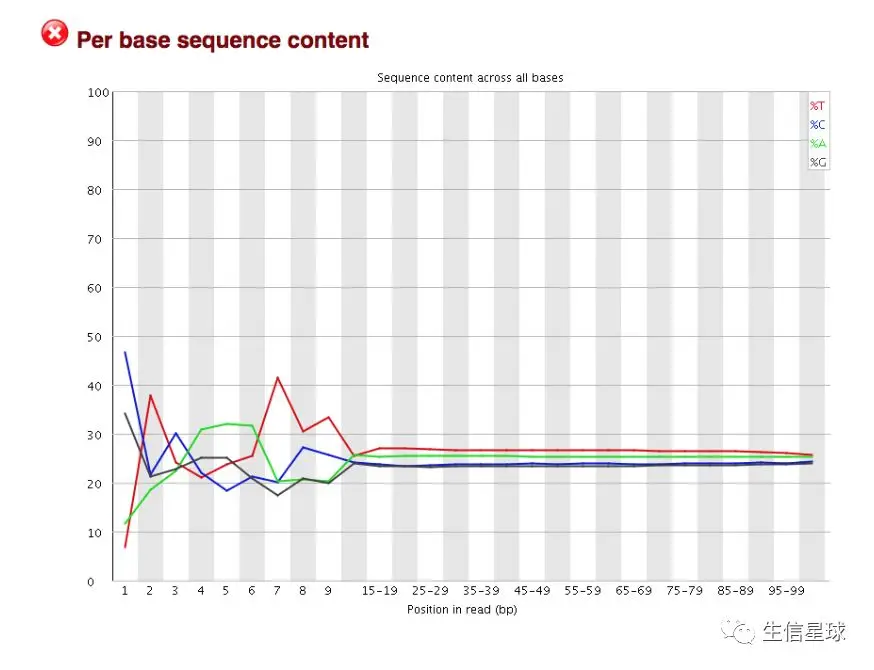
这个图经常会蹦出来FAIL 字眼吓唬我们。因为前10-12bp的碱基是RNA测序文库制备时使用的随机六聚体引物( ‘random’ hexamer priming) 随机引物是人工合成的随机序列六核苷酸混合物,这些引物可以随机地与 mRNA的任何部位互补,其优点是容易合成完整的cDNA
如果真的是测序随机的话,那么根据A-T配对、G-C配对,就可以得到每个位置的A和T比例应该差不多,GC比例应该差不多。但由于六聚体引物的存在,而且它也并不是真正的"随机”,还是存在一些碱基偏好性的,因此前10-12bp会有较大的波动
我们只要通过这个图,能看出没有特别大的碱基偏好性就好(也就是除了特殊的六聚体引物以外,A-T或C-G的比例差在1%以内就可以)
2.5 GC含量分布图 Per sequence GC content
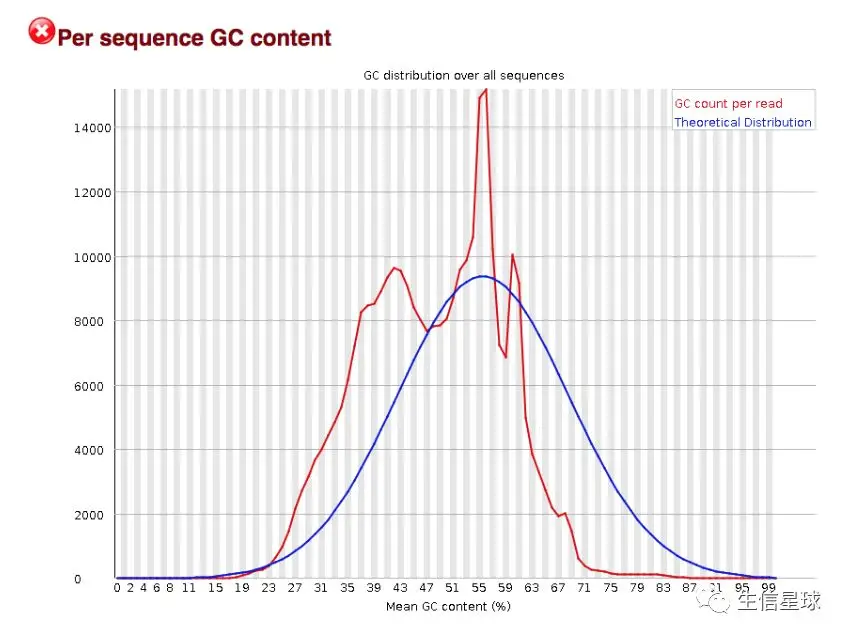
这个图表示了所有reads的GC分布,一般将中间的峰值和这篇文章(https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2909565/)列出的结果进行对比。
这个图理论上应该符合正态分布(也就是钟形曲线),除非有过表达的序列( over-represented sequences)[也就是在正态分布的基础上有一个尖尖的峰],或者存在其他物种的污染[也就是多个峰]
从这个图中可以看到,这个应该是符合过表达序列的情况,说明要么存在序列污染,要么是有个特别高表达的基因
2.6 重复序列数 Sequence Duplication Levels
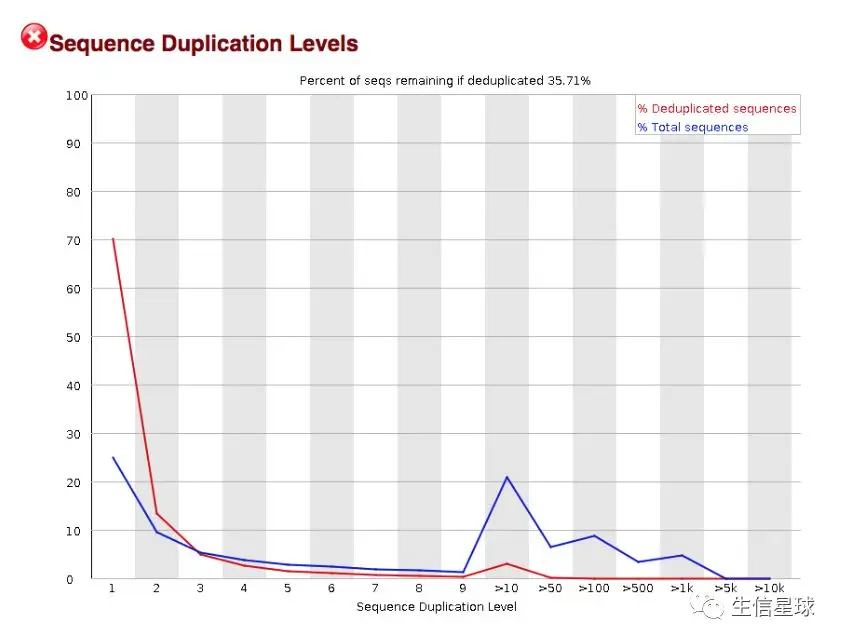
这个图可以帮助判断文库的复杂程度,如果PCR扩增次数太多或者起始扩增底物太少,都会降低文库的复杂度。
这个图中可以看到,似乎有大量的重复序列,也就是说文库复杂程度低,可能与某个基因的过表达有关
2.7 过表达序列表 Overrepresented sequences
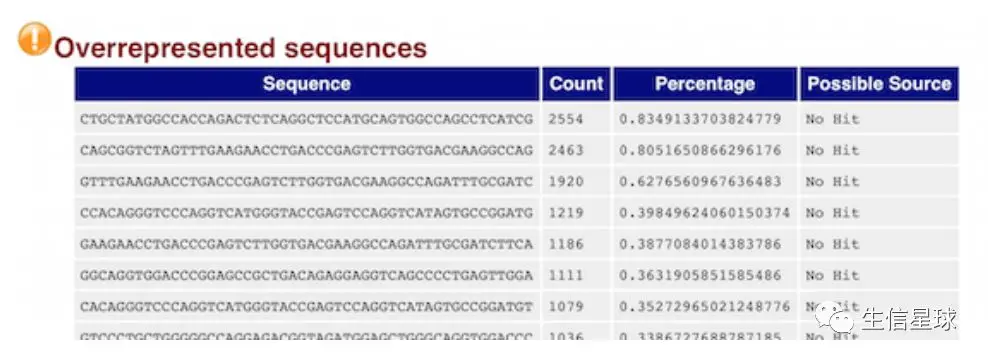
这个表的作用也非常重要!
它展示了长度至少20bp,数量占总数0.1%以上的reads碱基组成,它可以帮助判断污染(比如:载体、接头序列)
如果上面的GC含量分布图"挂了”,这个表可以帮助我们判断来源,如果是已知的载体或者接头,它会列出来;如果不是,可以复制序列去blast。
比如这里就可以去复制表达最多的第一条序列去blast,然后发现它其实是一个基因,于是可以验证之前的猜想:基因过表达