259-不同工具做的GO分析差异很大?不要蒙圈,很正常!
刘小泽写于2022.7.16 最近在比较同一个基因集使用不同工具进行GO分析时发现,即使使用的参数相同,结果可能也是差异很大
0 前言–举个例子
使用Gene Ontology的示例数据
id = c("APOH", "APP", "CND2", "COL3A1", "COL5A2", "CXCL6", "FGFR1",
"FSTL1", "ITGAV", "JAG1", "JAG2", "KCNJ8", "LPL", "LRPAP1", "LUM",
"MSX1", "NRP1", "OLR1", "PDGFA", "PF4", "PGLYRP1", "POSTN", "PRG2",
"PTK2", "S100A4", "SERPINA5", "SLCO2A1", "SPP1", "STC1", "THBD",
"TIMP1", "TNFRSF21", "VAV2", "VCAN", "VEGFA", "VTN")
0-1 使用Panther
Panther是GO指定的分析工具,分析的如何这里不做评价,只是来看看结果:
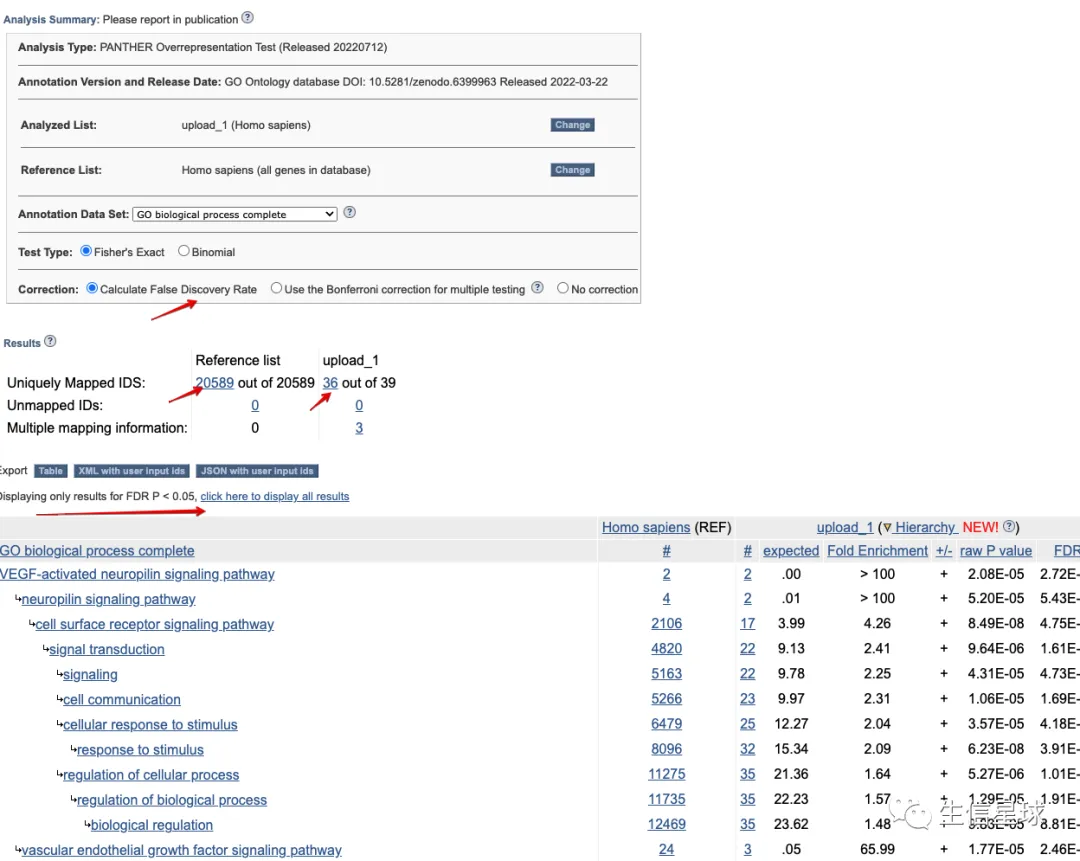
最后拿到了280个terms
0-2 使用clusterProfiler
同样使用FDR校正p值,然后阈值设为0.05
library(org.Hs.eg.db)
library(clusterProfiler)
ego <- enrichGO(gene = id,
OrgDb = org.Hs.eg.db,
ont = "BP",
pAdjustMethod = "fdr",
keyType = 'SYMBOL',
pvalueCutoff = 0.05,
qvalueCutoff = 0.05)
ego <- as.data.frame(ego)
最后拿到了357个terms
0-3 做个图
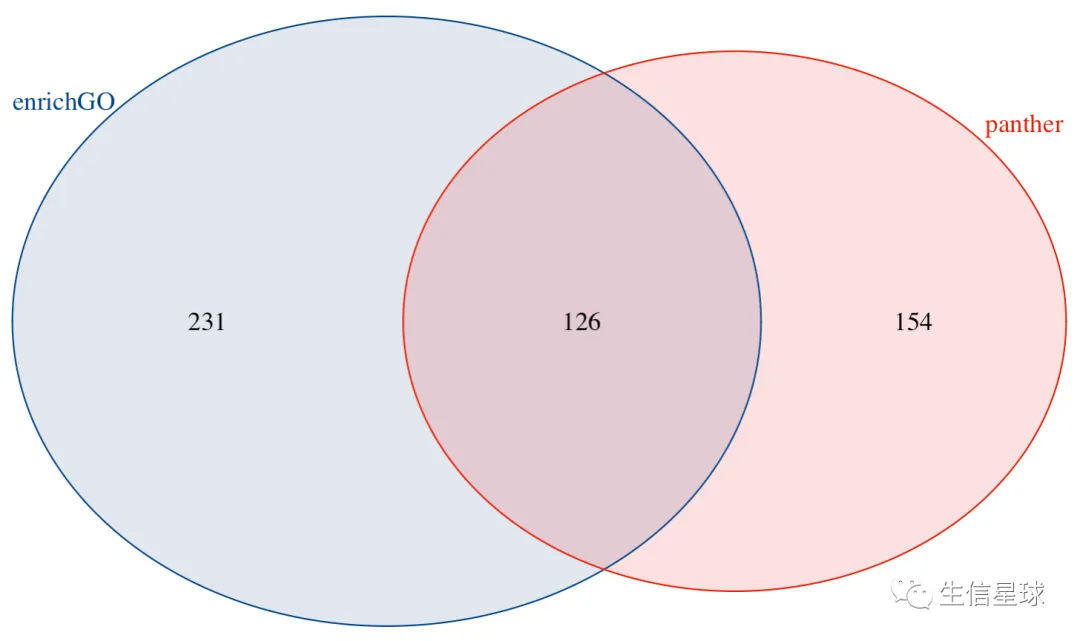
看到这个结果,你的内心可能会有如下波澜:
- 【惊讶】怎么差异这么大,两个都很流行并且都发表了文章,我应该相信哪个?
- 【怀疑】是自己的数据不正常吗?别人的会不会也是这样?
- 【躺平】算了,不管了,就用中间的交集部分也行
1 正式内容,推荐给你
偶然看到biostar有人写了一篇文章(Why does each GO enrichment method give different results?:https://www.biostars.org/p/122893/),写的不错,我把其中的关键点摘出来,和你分享。
1-1 首先什么是GO
- GO是一种描述性语言,而不是数据:GO is a means of providing consistent descriptions of genes/gene products between various databases and projects. GO is a language, not data.
- 你所认识的GO其实是基于它的基因注释集:Annotation sets of genes from a species with terms from GO are the data you need.
1-2 GO和Panther
Panther是分析工具,它不是GO。只是被GO官网作为一个展示基于GO数据的计算方式。因此,即使是同一种基因集,不同工具得到的结果大概率不同:PANTHER isn’t GO, it is a totally different set of annotations
1-3 既然都是基于基因集,那么不同工具使用的基因集一样吗?
这里列举了2个工具的说明,很明显,每个工具都有各自的基因集选取标准,谁优谁劣这里没做比较(并不是没有办法比较,应该也会有一些评测文章做这件事),只能说根据个人爱好去选择
- AgriGO: Raw GO annotation data is generated using BLAST, Pfam, InterproScan by agriGO or obtained from B2G-FAR center or from Gene Ontology.
- BinGO: Download the most recent annotation and ontology files from the GO website. You can use these as custom annotation/ontology files(http://www.psb.ugent.be/cbd/papers/BiNGO/Customize.html)
1-4 这些工具拿着GO注释数据,做了什么?
简单来说,注释数据千变万化,但核心还是分析方法:The GO annotations are simple curations, GO enrichment is a totally different thing.
或许从一开始的参数和检验方法,就注定了结果的差异:This is where the specific algorithm and parameters each tool is using really start to matter.
相信大家都遇到过,改变一个参数,就能得到不同的注释结果:Are you getting 17 instead of 213 because some parameter is different. Or, is it just because they’re different algorithms?
1-5 不要指望”异曲同工“
- Never assume that just because two algorithms claim to solve the same problem that you will get the same results.
- They’re doing different things, making different assumptions, require different input data and so on.
- Also, the semantics really matter, what each algorithm is actually telling you can be different.
1-6 分析结果可以有差异,但自己要做到心中有数
首先,自己要对自己的input数据有信心;
其次,不要被大量的富集结果的”胜利喜悦“冲昏头脑,因为有可能其中很多假阳性:Maybe the program that returned 213 results didn’t account for p-value or fold change in your differential expression. Are they all very strong or just a few?
很多时候,有几个重点关注的基因,富集到了理想的通路,这样才算分析成功。
1-7 不要被工具画的图所”迷惑“
很多时候,你可能会被不同工具画的图所吸引,以至于为了好看的图,不顾及数据是否合乎本身需求(当然,很多好的工具会同时注重二者)
- What the algorithm is doing is the important thing
- Never judge the performance of an algorithm based on the plots the programmer makes with the algorithm’s output
- You can always make pretty plots with good data, but a pretty plot with bad data is just bad.
- Find the tool that gives you the best quality results, deal with the plots later.
1-8 千万别忘了注释数据的”生产日期“
既然是数据,就是电子化的,随时有被淘汰的可能。为了得到更为准确的结果,比较推荐使用常常更新的数据库或者工具去进行分析(之前DAVID的坑,大家也不是不知道):always look at version history and release dates
1-9 关于GO和GOA
GO是形容词,比如可以描述:”a是b的子集“,”A等于B“,但GOA (annotations )是利用GO拿到的一些准确的名词,比如前面的a、b、A、B。
GO is basically adjectives, the annotations (GOA) are applying adjectives to nouns. The difference between gene ontology (GO) and gene ontology annotations (GOA) is critical. GO is standard, but GOAs are not.
举个例子
比如你想评价实验室的学生们,设置了几个形容词等级:优秀、良好、及格、不及格,其中给到学生A一个良好;但另一位PI使用同样的等级,对学生A提出了:不及格。那么这种:使用相同形容词,但由于各自判断标准不同,而导致结果差异的现象,就是GO和GOA的差异。
GO is what is called a controlled vocabulary, it provides a structured list of possible terms for genes. What GO does not do is regulate the means in which a gene gets annotated with a term.
再回到现实,Panther也能基于GO的描述去注释一些基因集,但它的评价标准和其他数据库的可能又不同,因此导致最后使用的基因集有差异。Panther比较好的一点是:它有自己的蛋白家族本体库,它能对GO进行扩展
那么官方的GOA如何产生呢?
GO consortium generates their “reference” GOAs: http://www.ncbi.nlm.nih.gov/pubmed/19578431
这篇文章就解释了以下3个问题:
- For one, pubs and documents online are where you find details of how they built their annotations and what exactly they’re using for a vocabulary/ontology (GO only, GO + Extra, GO Slim, how old is the GO version?)
- Second, this will tell you how they came up with the GOA, which is important?
- Finally, even if two different enrichment tools use the same GOA set, they’ll come up with different results
了解了GO和GOA的区别,你是不是对不同工具的不同基因集导致差异,有了一定的认识呢?
1-10 再次看个例子
这4组分析者同时对一组基因采用不同策略,他们各有各的道理
- Group A may have used a less strict method to annotate genes
- Group B may have only annotated genes where experimental evidence was present
- Group C could have taken a hybrid approach
- Group D could have done the same as C but with different tools.
我们要做的是:sanity checks (e.g. checking p-values/fold changes against enrichment sets)
因为大部分发表的工具,方法肯定都是经过检验的,但通过检查,我们可以判断:which one is too strict (sets are very small, false negatives) or it isn’t strict enough (large sets, false positives).
目前的现实问题是:如果你提交的结果中,存在很多低表达的差异基因或者蛋白,然后使用的ORA方法(当然GSEA对整体表达的考虑会优于常规的ORA方法,这里不对GSEA做讨论)拿到了很多的基因集。
审稿人就会提出疑问:真的会富集到这么多吗?你会不会使用过于宽松的条件,或者使用的是一些偏门或者过时的基因集呢?
1-11 常见的分类
目前市面上的分析工具可能会分成这么几类:
- some tools use different algorithms to calculate enrichment
- some exist simply because they produce cute figures
- some tools are a means of getting data from multiple ontologies/pathway/etc annotations
他们或许基于不同的基因集,比如开头提到的Panther是基于GO Ontology database DOI: 10.5281/zenodo.6399963 Released 2022-03-22,clusterPrfilter基于Biocondutor的GO.db 和org.db(伴随着Bioconductor半年更新一次,目前Bioconductor是v3.15 2022-03-10)
但好的工具应该具备的条件是:处理假阳性
- Some tools have very good corrections, other tools don’t. Check which tools are making corrected p-values and what methods they use.
- You may have seen 17 instead of 213 because the tool with 213 genes in one cluster didn’t make any or used a less strict method for p-value corrections.
1-12 忠告
- never “fire and forget”
- always check results from different programs
- try different settings
- be sure you understand what each program is really doing
- always spot check your results against your data
湿实验如此谨慎,为何对干实验这么放松警惕? You’d never run a western to test a new antibody without controls for cross reactivity and a known working antibody, don’t run bioinformatics programs blindly.