214-如何使用DiffBind进行peaks的差异分析?
刘小泽写于2020.11.20 在使用call peaks工具(如MACS2等)对bam文件操作,找到peaks文件后,有时还想探索一下两个不同处理的样本之间peaks有没有差异。注意:call peaks是每个处理样本与input之间比较(可能有时还没有input);而diffbind做的是将每个处理样本与input之间比较完后,再比较处理之间各自的peaks差异
前言
官方文档:https://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf
如果之前安装的V2版本,很多函数都不能用,比如dba.normalize();并且V2的示例数据与官方文档中也是对应不起来的
但是要安装V3版本,还稍微有点复杂:
# 首先需要使用R 4.0版本,其次需要Bioconductor的3.12(3.11都不能安装DiffBind的V3)
BiocManager::install(version = "3.12")
# 如果之前有V2版本,可以卸载掉
detach("package:DiffBind", unload=TRUE)
remove.packages('DiffBind')
# 检查版本
suppressMessages(library(DiffBind))
> package.version('DiffBind')
[1] "3.0.6"
DiffBind是对peaks文件进行操作的,那么什么叫做peaks文件呢?
简单说,peaks文件就是表示各个处理的蛋白结合的基因组区域,是BED格式的,即包括染色体、起始和终止,有时还会补充上一个分值表示peak的强度或者可信度
一般这个软件会有5步:
- 根据meta信息进行peaks文件的读取:可以是ChIP-Seq peak caller(比如MACS2)得到的结果,或者自己设置的基因组区域或者基因组上所有的启动子区域,反正格式要符合。可以接受csv格式,或者xls、xlsx格式;另外,如果一个样本有多个方法得到的peaks文件,是可以一起放在meta信息中的
- 对peaks的质控:比如看看组间或组内的相关性、同一个条件下不同的peak caller结果差异大不大;还可以根据blacklist对peaks进行过滤
- 统计reads数:一旦有了比较高质量的peaks区间,就可以根据bam来计算这些区间内的reads数
- 【核心】差异分析:默认使用deseq2,根据各个样本的peaks得到的reads数进行差异分析,和转录组类似,也是需要构建一个分组矩阵。
- 绘图与结果导出:可以绘制MA plot、火山图、热图、PCA、箱线图;结果也会给出differentially bound sites的位置,方便后面的注释、motif寻找等等
快速上手
注意下面出现的所有DBA函数的意思都是:Differential Binding Analysis
0 加载测试数据
这个数据是5个乳腺癌细胞系的ERa转录因子ChIP结果,其中有3个细胞系对它莫西芬(一种抗雌激素)有反应(Responsive),两个有抵制作用(Resistant)。每个细胞系都至少有2个重复,有一个还有3个重复,所以总共有11个MACS2 call peaks结果。测试数据中只选取了chr18的数据
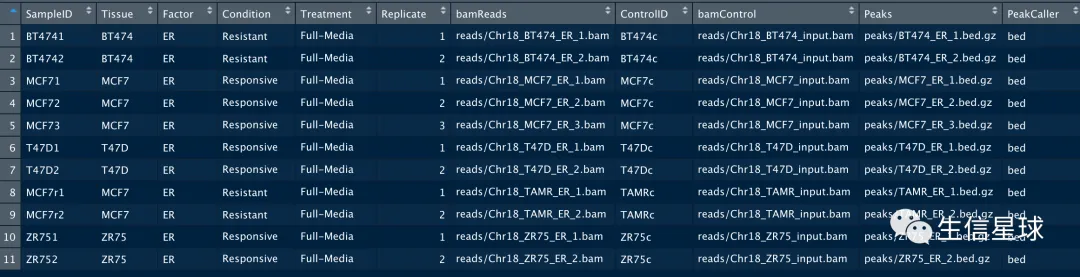
file=file.path(system.file("extra", package="DiffBind"),"tamoxifen.csv")
samples=read.csv(file)
> names(samples)
[1] "SampleID" "Tissue" "Factor" "Condition"
[5] "Treatment" "Replicate" "bamReads" "ControlID"
[9] "bamControl" "Peaks" "PeakCaller"
之后应用到自己的数据,也是要准备一个这样的表格,其中需要有bam、call peaks的结果
1 读取数据
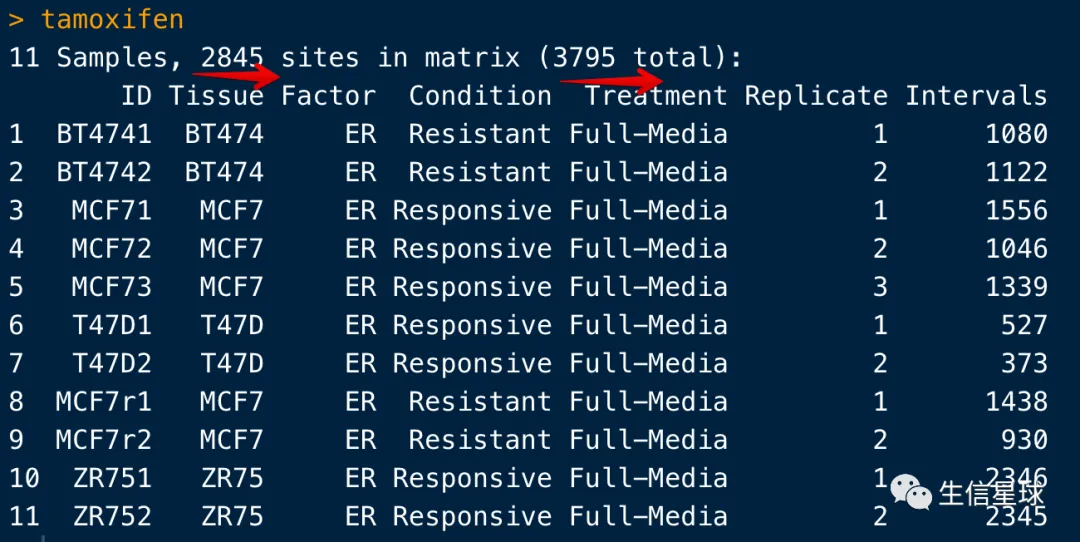
tamoxifen=dba(sampleSheet="tamoxifen.csv",dir=system.file("extra", package="DiffBind"))
# BT4741 BT474 ER Resistant Full-Media 1 bed
# BT4742 BT474 ER Resistant Full-Media 2 bed
# MCF71 MCF7 ER Responsive Full-Media 1 bed
# MCF72 MCF7 ER Responsive Full-Media 2 bed
# MCF73 MCF7 ER Responsive Full-Media 3 bed
# T47D1 T47D ER Responsive Full-Media 1 bed
# T47D2 T47D ER Responsive Full-Media 2 bed
# MCF7r1 MCF7 ER Resistant Full-Media 1 bed
# MCF7r2 MCF7 ER Resistant Full-Media 2 bed
# ZR751 ZR75 ER Responsive Full-Media 1 bed
# ZR752 ZR75 ER Responsive Full-Media 2 bed
- 第一行的2845表示:至少两个样本中有重叠的peaks数量(overlap in at least two of the samples);
- 第一行的3795表示:所有的重叠peaks放在一起并去重后的数量(total number of unique peaks after merging overlapping ones)
可以简单画个图:
plot(tamoxifen)
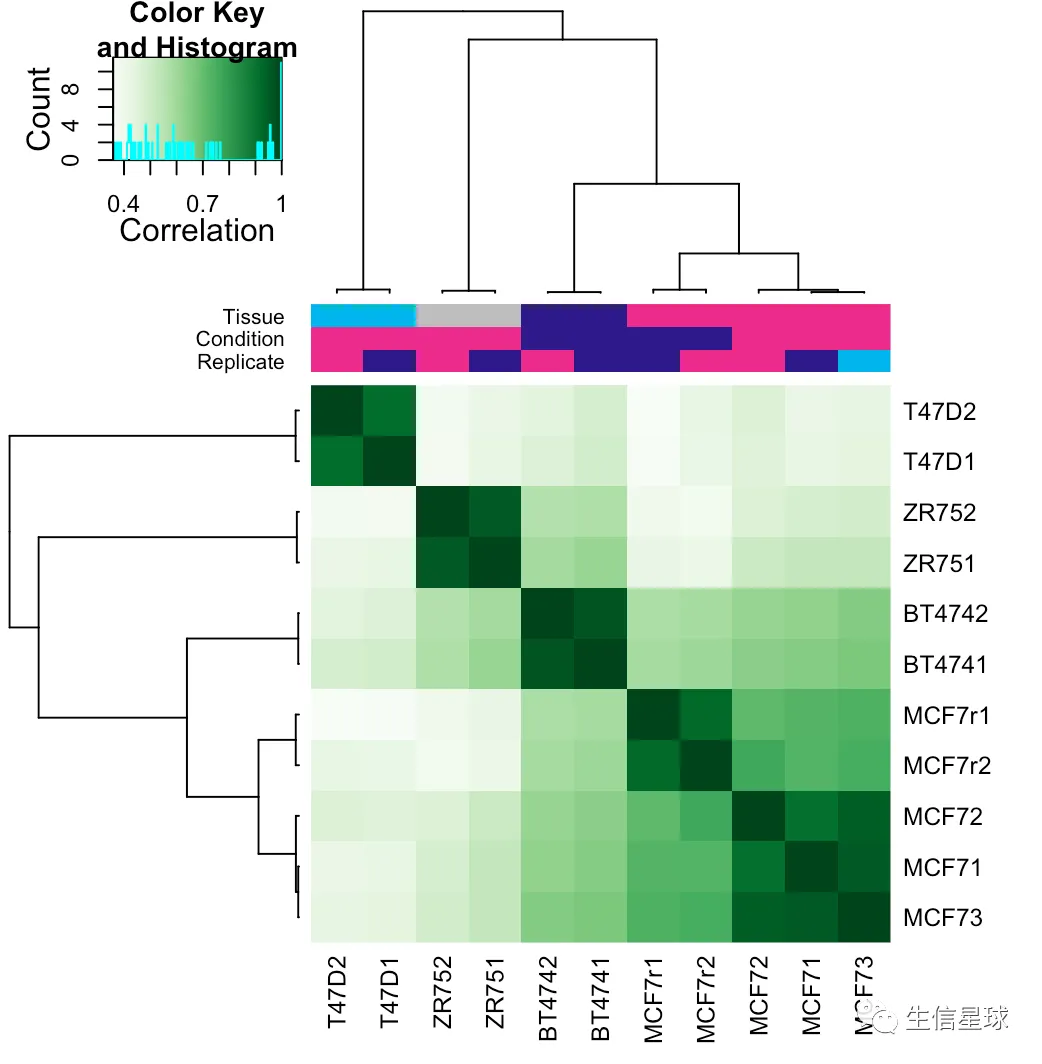
图中可以看到:
- 两组MCF7细胞系的组内相关性最高;
- 不同的condition(Responsive vs Resistant)还是有差别的:responsive (MCF7, T47D, and ZR75) vs. resistant (BT474 and MCF7r)
2 关于Blacklists
一般来说,分析ChIP-Seq数据时,考虑Blacklists是个好习惯。
是什么?
基因组上会存在一些特定的重复序列,例如在丝粒、端粒以及卫星重复序列,特点就是重复序列区域的碱基完全相同。而二代测序的数据进行比对时(比如现在有重复区域A和B,而A和B的碱基完全相同),仅仅依靠比对的算法,是不能判断reads比对到A还是B的。这时不同的软件会进行判断:有的软件会随机选取一个,有的软件会两个区域都进行计算。这种计算上的不确定性导致了这些区域的测序深度普遍偏高。
回归ChIP-Seq数据,我们一般是通过比较IP组和input组之间基因组上测序深度的差异,通过这个差异再加上一些计算方法,macs2等软件就会帮我们得到一系列peaks,也就得到了结合位点附近区域。如果我们是通过测序深度来确定peaks,那么重复序列区域的虚高情况势必会造成影响。因此,这部分区域被列入了“黑名单”。
因此,Blacklists指的是:根据参考基因组挑出来有问题的一些区间,比如ENCODE就提供了这样的文件,并区分了物种和基因组版本。这里可以用dba.blacklist来调取
怎么做?
data(tamoxifen_peaks)
tamoxifen
# 看看现在的peaks数量
peakdata <- dba.show(tamoxifen)$Intervals
# 去除以后
tamoxifen <- dba.blacklist(tamoxifen, blacklist=DBA_BLACKLIST_HG19,
greylist=FALSE)
# 去除以后的数量
peakdata.BL <- dba.show(tamoxifen)$Intervals
# 看看每个样本去除了多少的blacklist区域peaks
peakdata - peakdata.BL
# [1] 0 0 1 1 1 0 0 0 0 0 0
# 可以看到,这里只有中间的三个样本(MCF7)去掉了1的peaks
3 统计reads数
这里就需要用到bam文件了,正常情况下,使用
tamoxifen <- dba.count(tamoxifen)
就会得到结果
只是这里示例数据已经给出,直接调用即可
data(tamoxifen_counts)
tamoxifen_counts
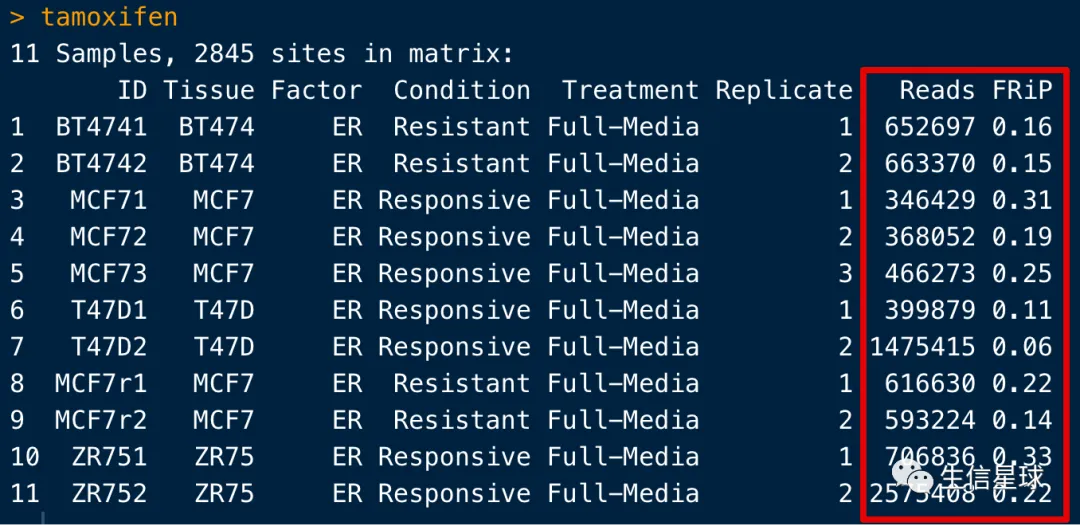
看到这里比之前多了两列:第一个是每个样本中所有比对上的reads数量;第二个是FRiP(Fraction of Reads in Peaks):也就是该样本peaks上的reads占所有reads的百分比,可以用来展示这个样本富集的效果。比如可以看看:
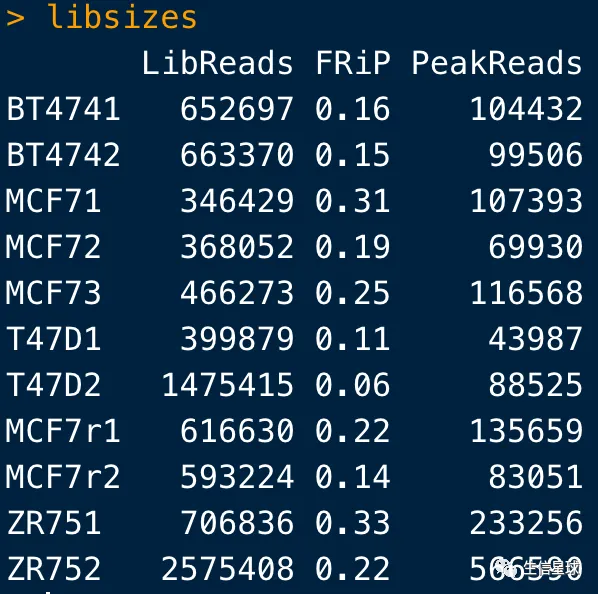
info <- dba.show(tamoxifen)
libsizes <- cbind(LibReads=info$Reads, FRiP=info$FRiP,
PeakReads=round(info$Reads * info$FRiP))
rownames(libsizes) <- info$ID
libsizes
当然,统计完后,为了后面差异分析的可靠性,还是需要进行标准化的(就像RNA-Seq的normalization去除文库大小差异)
# 这里默认也是基于测序深度进行normalize
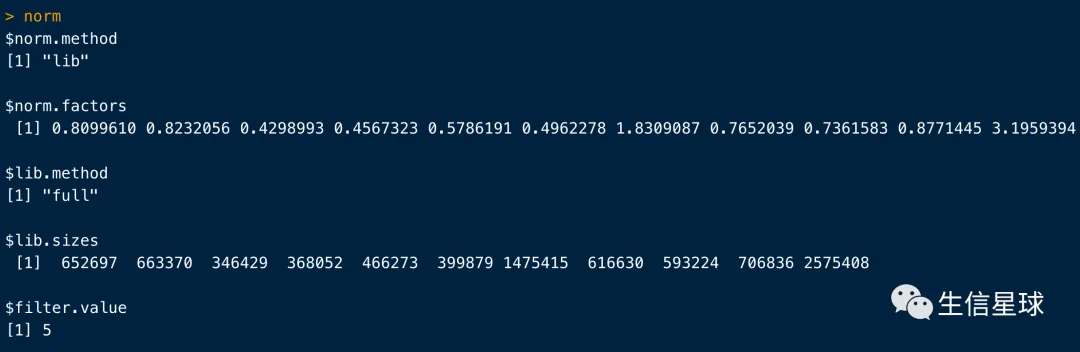
tamoxifen <- dba.normalize(tamoxifen)
# 可以看一下具体的细节
norm <- dba.normalize(tamoxifen, bRetrieve=TRUE)
norm
可以看到包括使用的方法(基于lib即文库大小),计算的normalization factor,总的文库大小等
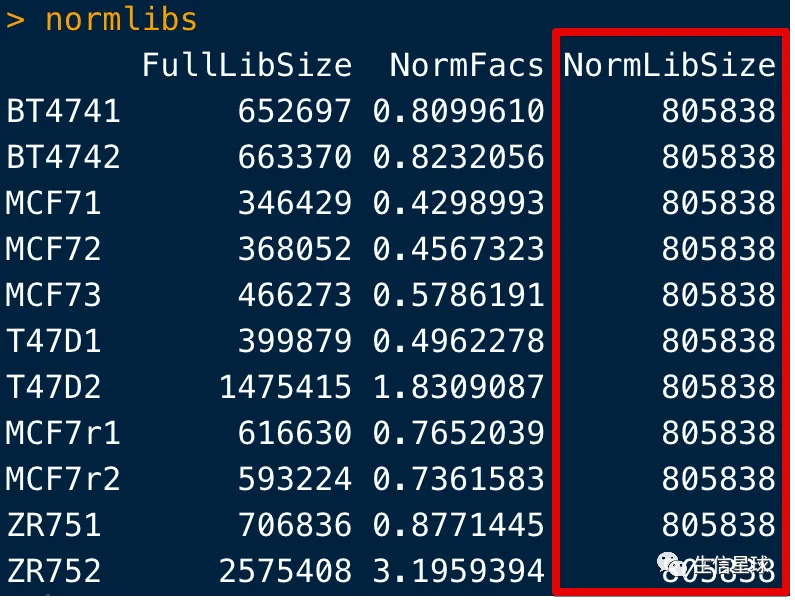
如果用norm.factors乘以lib.sizes,就会得到标准化后的文库大小
normlibs <- cbind(FullLibSize=norm$lib.sizes, NormFacs=norm$norm.factors,NormLibSize=round(norm$lib.sizes/norm$norm.factors))
rownames(normlibs) <- info$ID
4 构建比较矩阵+差异分析
还记得limma的三大矩阵吗?
- 表达矩阵:就是熟知的matrix形式
- 分组矩阵:也就是实验设计(design)矩阵
- 差异比较矩阵:就是说那几个组之间进行比较
默认使用deseq2,不过这里也是需要设置谁和谁进行比较
tamoxifen <- dba.contrast(tamoxifen,
reorderMeta=list(Condition="Responsive"))
这里将Responsive设为“对照”,当然这也是最简单的设置方式

更为复杂一点的是,同时考虑多个因素:
比如看到上图中,虽然都是MCF7细胞,但处理表现也是不同的,既有resonsive也有resistant。于是把Tissue的信息加进来综合考量
tamoxifen <- dba.contrast(tamoxifen,design="~Tissue + Condition")
构建完比较矩阵后,就可以进行差异分析了
tamoxifen <- dba.analyze(tamoxifen)
dba.show(tamoxifen, bContrasts=TRUE)
# Factor Group Samples Group2 Samples2 DB.DESeq2
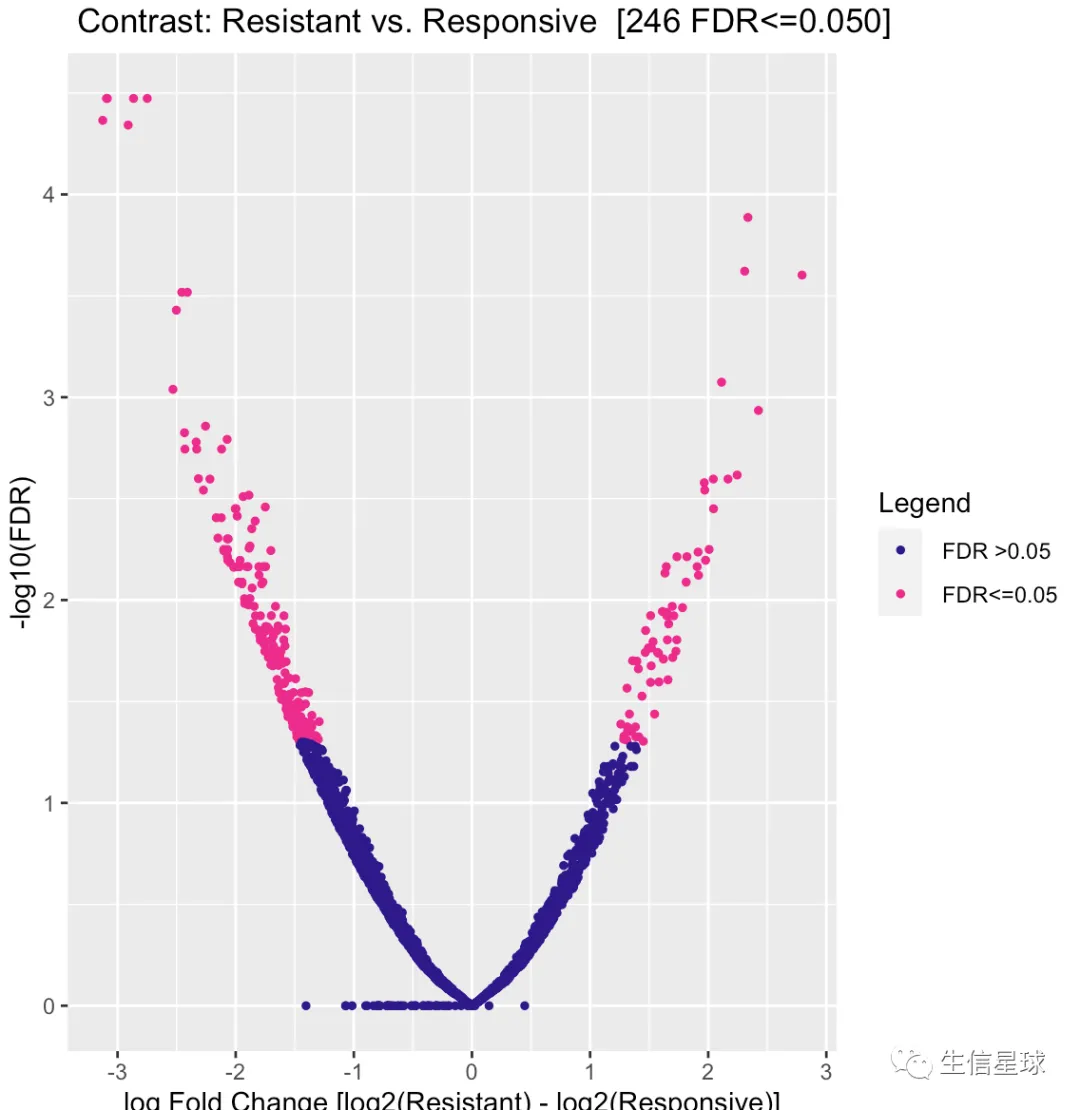
# 1 Condition Resistant 4 Responsive 7 246
看到结果说找到了246个differentially bound (DB)【默认阈值是:FDR <= 0.05】
当用这246个binding sites去做相关性,会看到和之前的结果有不同,更接近真实的情况:4个 tamoxifen resistant 样本更好地聚在了一起;而之前使用全部的2845个binding sites就看不到
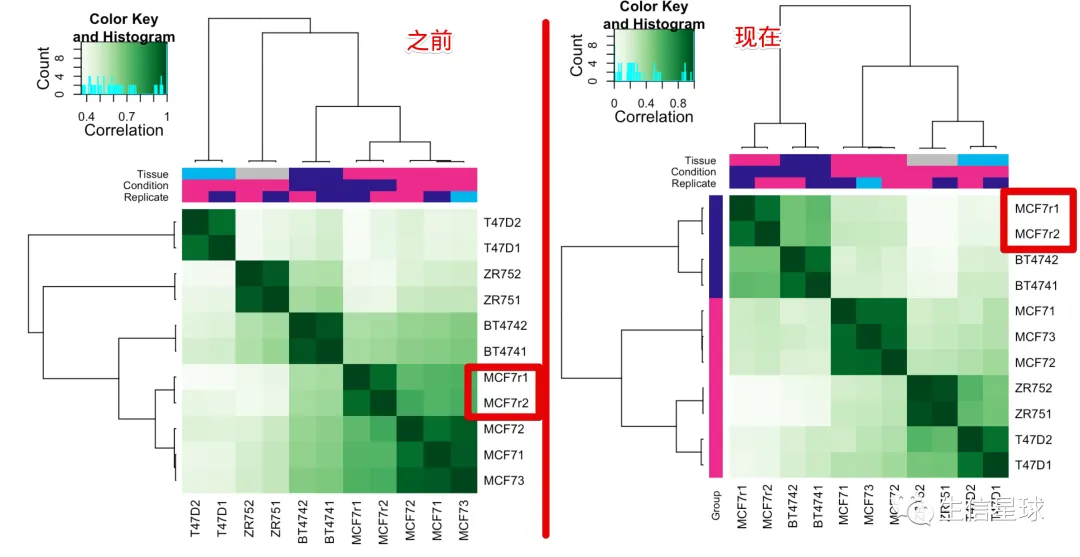
还可以提取出differentially bound sites
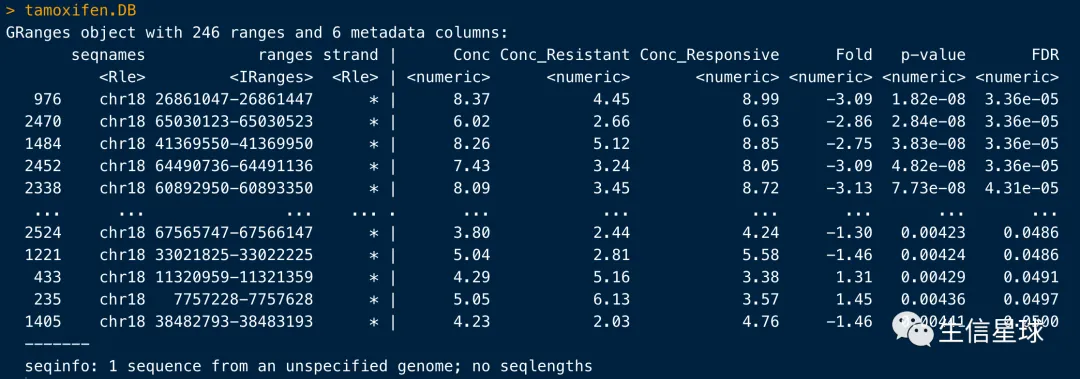
tamoxifen.DB <- dba.report(tamoxifen)
提取的是一个GRanges对象,其中:
Conc指的是Mean read concentration over all the samples (进行了log2的处理)Conc_Resistant指的是在所有resistant的样本中的统计;下一列同理Fold这里指的是Resistant/Responsive,为正数表示 Increased binding affinity in the Resistant group
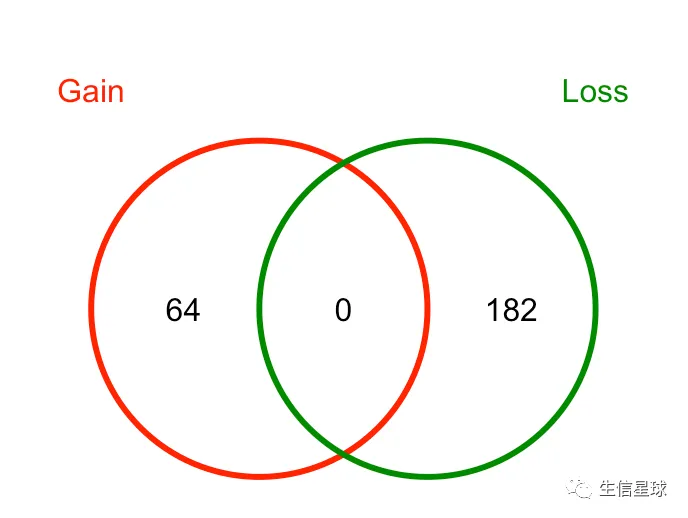
然后可以统计一下正负的数量
sum(tamoxifen.DB$Fold>0)
# [1] 64
sum(tamoxifen.DB$Fold<0)
# [1] 182
5 画图
5.1 venn 图
dba.plotVenn(tamoxifen, contrast=1, bDB=TRUE,
bGain=TRUE, bLoss=TRUE, bAll=FALSE)
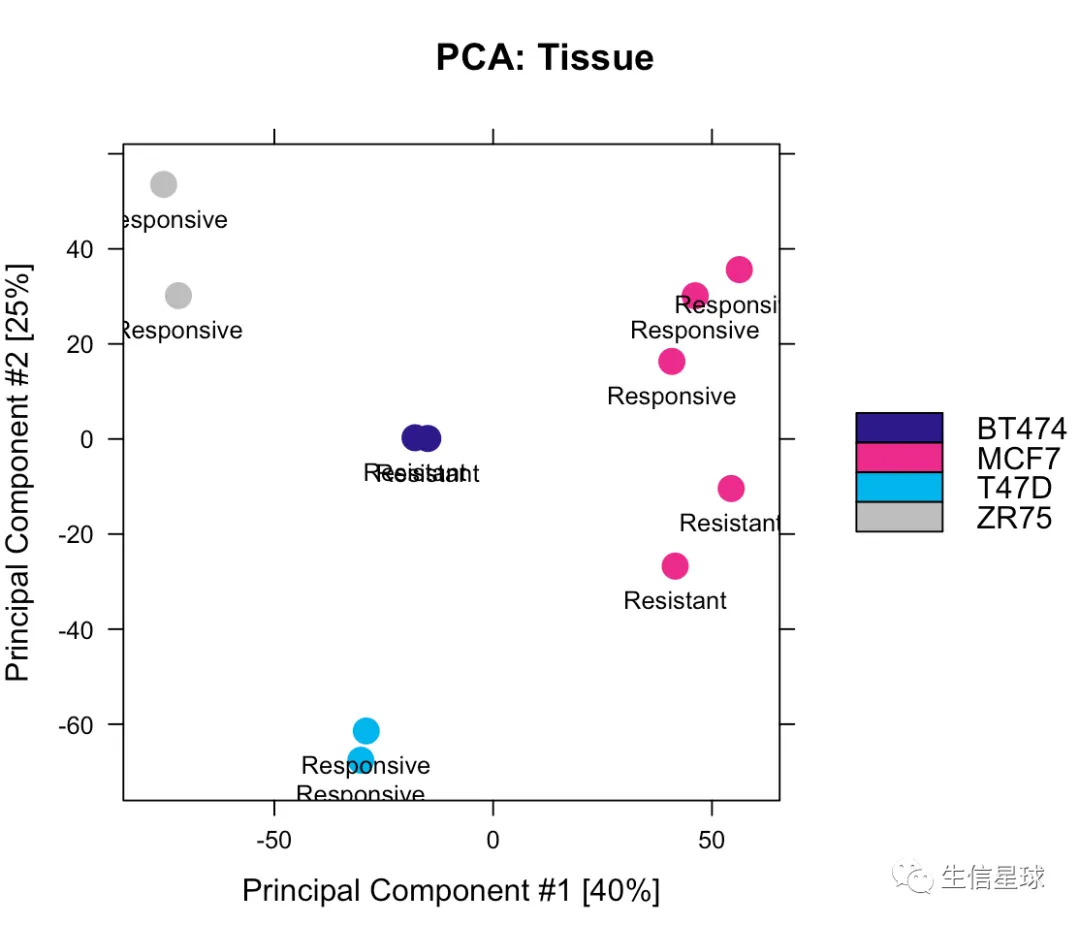
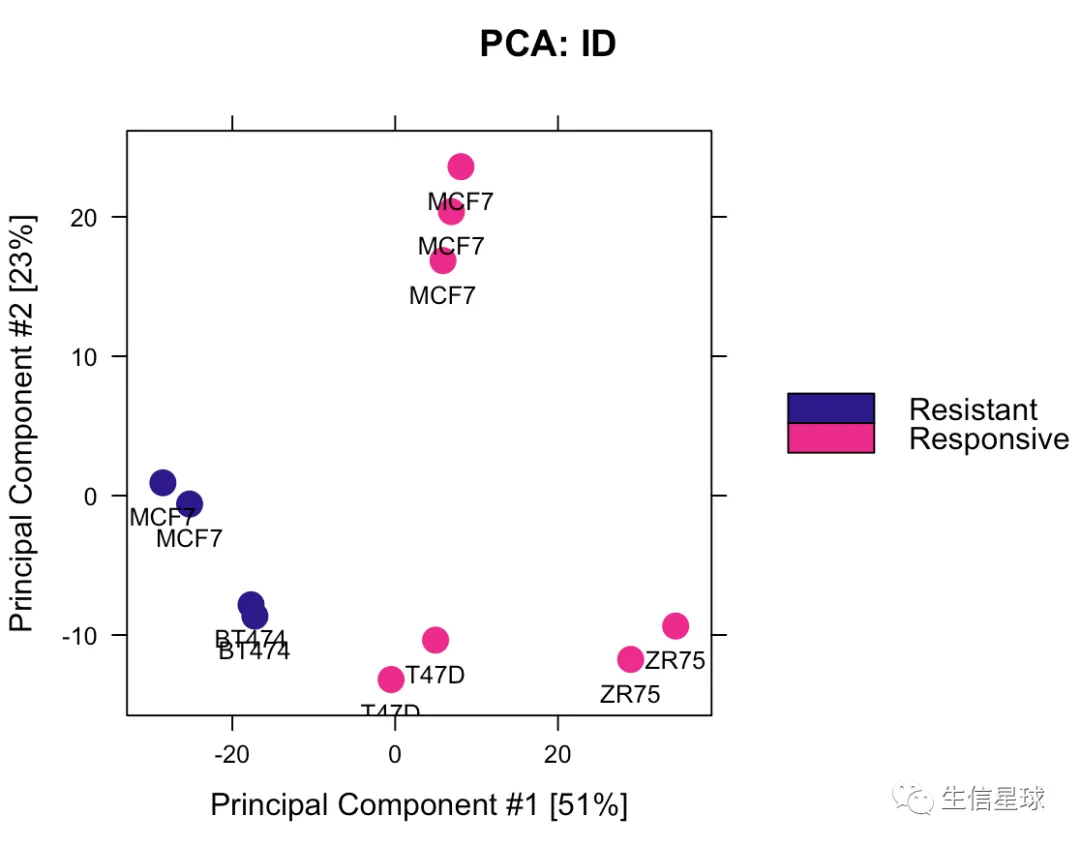
5.2 PCA
# 如果使用全部的2845 sites
dba.plotPCA(tamoxifen,DBA_TISSUE,label=DBA_CONDITION)
# 如果使用差异的246sites
dba.plotPCA(tamoxifen, contrast=1, label=DBA_TISSUE)
一个非常明显的不同就是MCF7分开了
还可以画3D版本,但需要安装rgl包
dba.plotPCA(tamoxifen,contrast=1, b3D=TRUE)
5.3 火山图
dba.plotVolcano(tamoxifen)
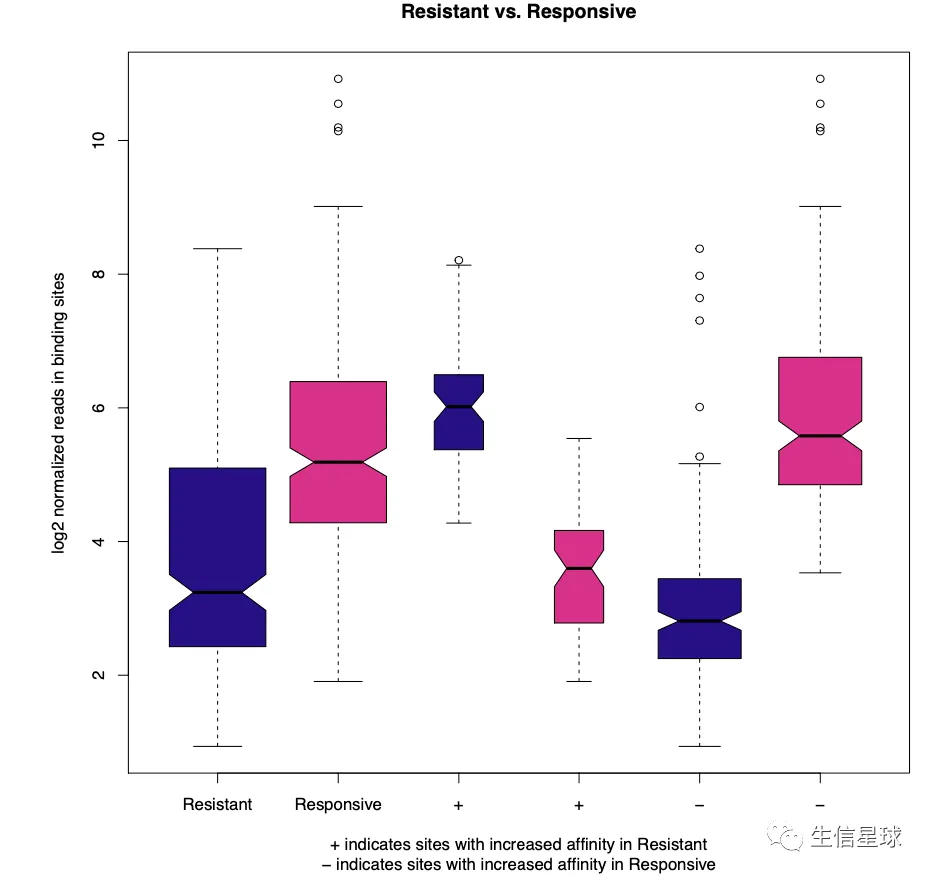
5.4 箱线图
pvals <- dba.plotBox(tamoxifen)
# 返回的结果是一个p-values的矩阵,根据two-sided Wilcoxon ‘Mann- Whitney’ test计算得到
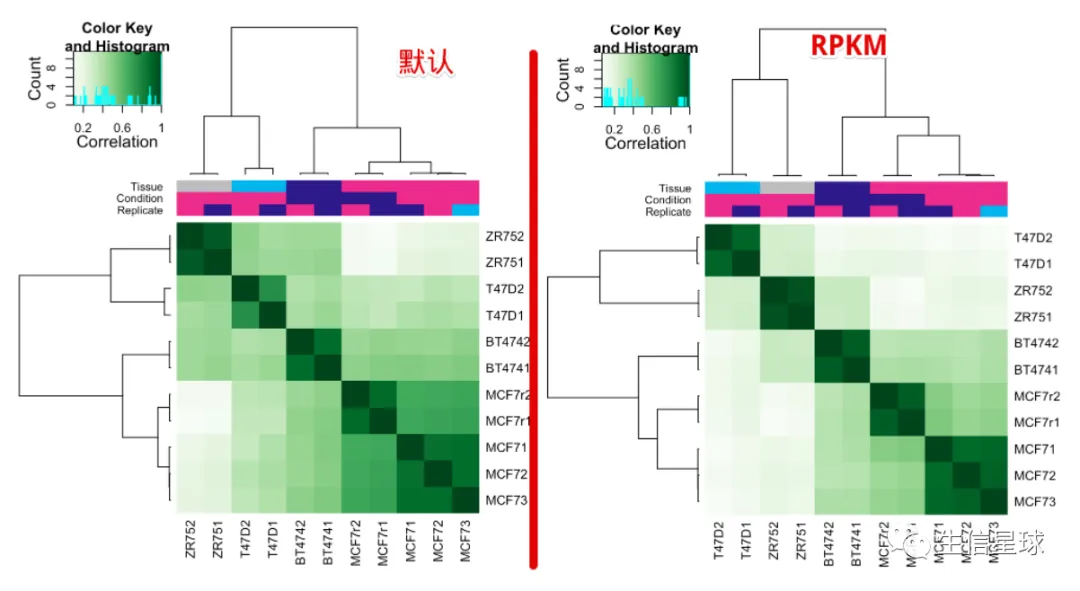
5.5 热图
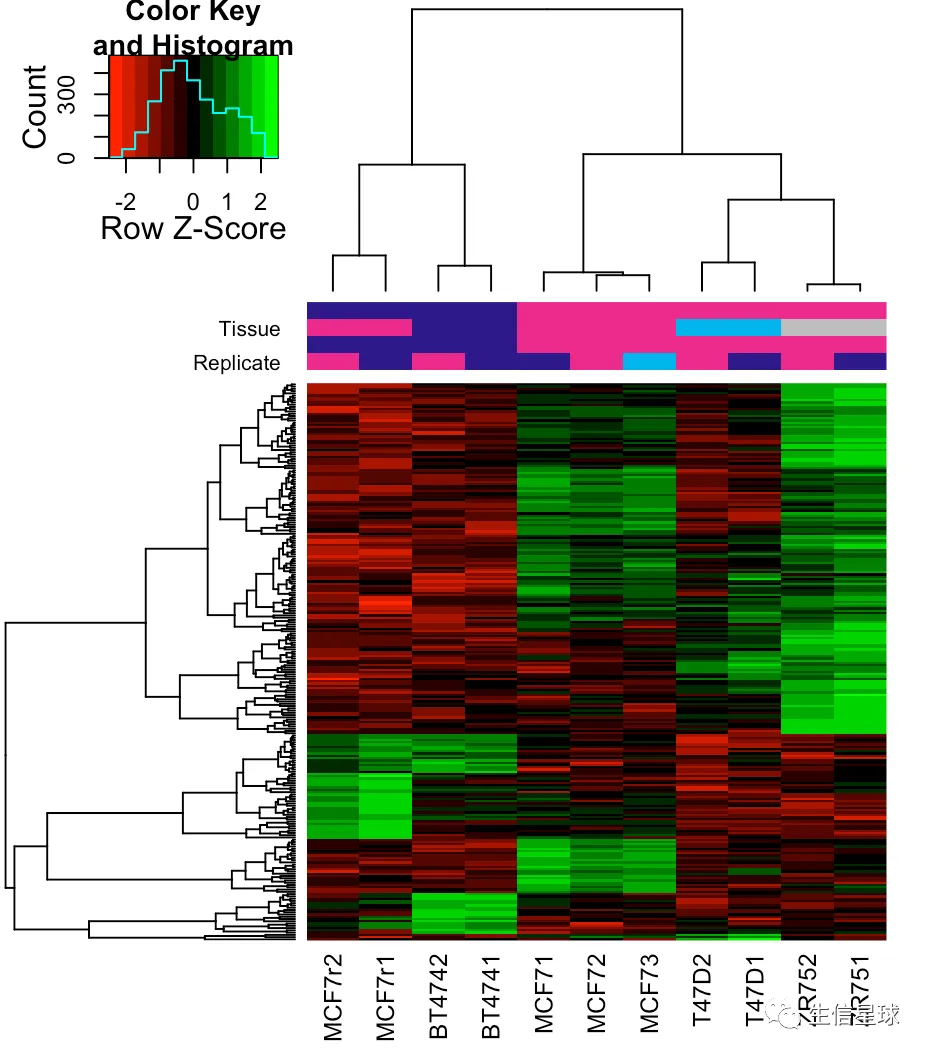
# 使用全部的sites
corvals <- dba.plotHeatmap(tamoxifen)
# 默认使用DBA_SCORE_NORMALIZED,还可以用另一种计算normalization score的方法:RPKM fold (RPKM of the ChIP reads divided by RPKM of the control reads)
dba.plotHeatmap(tamoxifen, score=DBA_SCORE_RPKM_FOLD)
# 如果只用差异的246sites
hmap <- colorRampPalette(c("red", "black", "green"))(n = 13)
readscores <- dba.plotHeatmap(tamoxifen, contrast=1, correlations=FALSE,scale="row", colScheme = hmap)