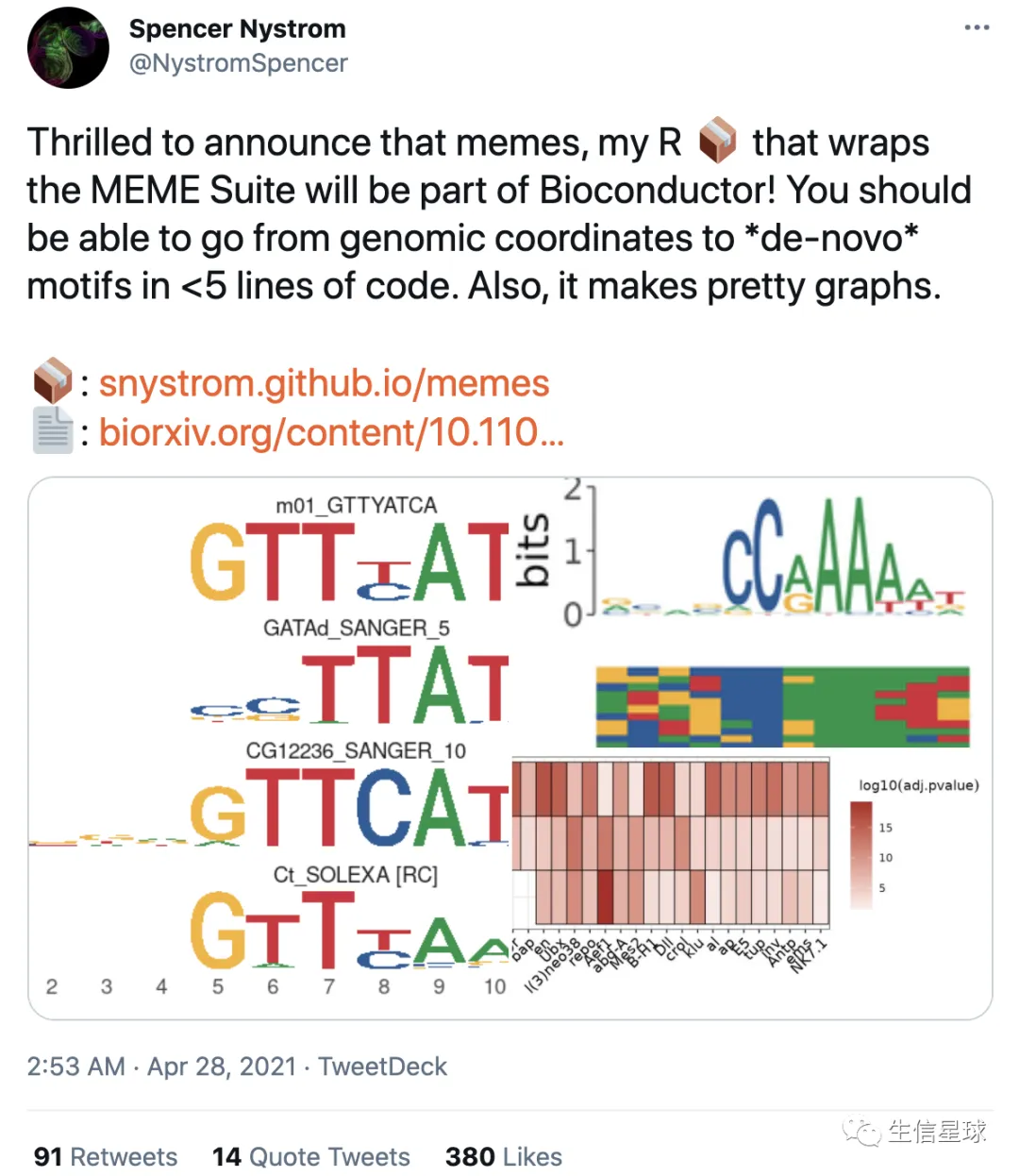
244-寻找motif,R版本的MEME来了,不试试?
刘小泽写于2021.4.28 会介绍MEME的用法,以及新的R版本使用,最后看看其他类似工具
什么是MEME?
看他的官网:http://meme-suite.org/index.html,很清楚写着基于motif进行的一系列分析。
在官网中找到的motif定义: A motif is an approximate sequence pattern that occurs repeatedly in a group of related sequences.
关键词:大量重复出现;短序列,只是一段pattern
当然,这段序列不局限于DNA,还可以是RNA或者氨基酸序列;并且它经常是具有序列特异性的结合位点(比如转录因子结合位点或者RNA剪切位点等)

左侧列表就写出了它的五大功能:
- Motif Discovery:预测序列上的motif信息,可以是DNA,RNA,氨基酸序列,方法是从头预测(de novo discovery)。比如我们ChIPseq数据拿到了一些peaks的区间,那么就能拿到序列,然后就能拿到序列中的motif
- MEME: Multiple EM for Motif Elicitation, 利用de novo预测得到motif结构和在输入序列上的位置
- DREME: Discriminative Regular Expression Motif Elicitation, 主要针对长度在100bp左右的序列
- STREME: Sensitive, Thorough, Rapid, Enriched Motif Elicitation,它是DREME的替代版,特点是基于的序列数量多(多于50个)
- MEME-ChIP
- GLAM2
- MoMo
- Motif Enrichment:分析已知的motif在我们的序列上有没有,有多少?
- CentriMo
- AME
- AME
- SpaMo
- Motif Scanning:分析我们序列上可能出现motif的位置是?
- FIMO
- MAST
- MCAST
- Motif Comparison:不同motif之间的相似性有多少?
- Tomtom
- Gene Regulation:预测基因和调控元件(用染色体区域表示)之间的调控关系

但它的缺点就是:它作为一个网页工具,但略显繁琐,限制了使用人群
新的R版本怎么用?
官方文档在:https://snystrom.github.io/memes-manual/
今天看到一个推文,发现有了R版本的支持,作者还发了篇文章:Memes: an R interface to the MEME Suite(https://www.biorxiv.org/content/10.1101/2021.04.23.441089v1)
In addition to interfacing with popular MEME Suite tools, memes leverages existing R/Bioconductor data structures to store the complex, multidimensional data returned by MEME Suite tools for rapid data access and manipulation.
首先安装R包
现在这个R包应该还是在开发版,需要R 4.1以上才可以。我于是下载了R 4.2和4.1安装试一下,结果失败
不过没关系,先把安装代码放在这里,毕竟过些日子出了正式版4.1就可以了 (R 4.1 scheduled for Tuesday 2021-05-18)
# 需要:Bioconductor version: Development (3.13)
# Bioconductor version '3.13' requires R version '4.1'
# 使用R 4.1 及以上版本
if (!requireNamespace("remotes", quietly=TRUE))
install.packages("remotes")
remotes::install_github("snystrom/memes")
R包需要本地MEME套件(for Linux, OS X and Cygwin systems)
不得不说,安装过程都这么繁琐,确实影响了使用者心情
# 下载, 在 https://meme-suite.org/doc/download.html/ 找版本
# 安装
tar zxf meme-5.3.3.tar.gz
cd meme-5.3.3
./configure --prefix=$HOME/meme --enable-build-libxml2 --enable-build-libxslt
make
make test
make install
# 最后添加环境变量
export PATH=$HOME/meme/bin:$HOME/meme/libexec/meme-5.3.3:$PATH
# 这个倒是安装成功了,耗时约10min
如果之后安装成功了,那么就继续运行:
先来一个检测步骤
library(memes)
check_meme_install()
#> checking main install
#> ✓ /opt/meme/bin
#> checking util installs
#> ✓ /opt/meme/bin/dreme
#> ✓ /opt/meme/bin/ame
#> ✓ /opt/meme/bin/fimo
#> ✓ /opt/meme/bin/tomtom
#> ✓ /opt/meme/bin/meme
# 或者可以指定路径
check_meme_install(meme_path = 'bad/path')
核心的函数有:
DREME寻找motif
suppressPackageStartupMessages(library(magrittr))
suppressPackageStartupMessages(library(GenomicRanges))
# 拿到peaks
data("example_peaks", package = "memes")
# 拿到基因组
dm.genome <- BSgenome.Dmelanogaster.UCSC.dm6::BSgenome.Dmelanogaster.UCSC.dm6
# Generate sequences from 200bp about the center of my peaks of interest
sequences <- example_peaks %>%
resize(200, "center") %>%
get_sequence(dm.genome)
# 最后运行dreme
# runDreme() accepts XStringSet or a path to a fasta file as input.
# In a real analysis, e should typically be < 1
dreme_results <- runDreme(sequences, control = "shuffle", e = 50)
# 展示结果
library(universalmotif)
dreme_results %>%
to_list() %>%
view_motifs()

TOMTOM比较motif
# Discovered motifs can be matched to known TF motifs using runTomTom()
# TomTom uses a database of known motifs which can be passed to the database parameter as a path to a .meme format file, or a universalmotif object.
# 可以指定数据库:
# options(meme_db = system.file("extdata/flyFactorSurvey_cleaned.meme", package = "memes"))
full_results <- dreme_results %>%
runTomTom()
# 可视化结果
full_results %>%
view_tomtom_hits(top_n = 1)

AME富集motif
# AME is used to test for enrichment of known motifs in target sequences
# 同样也是需要数据库的
# options(meme_db = "path/to/database.meme")
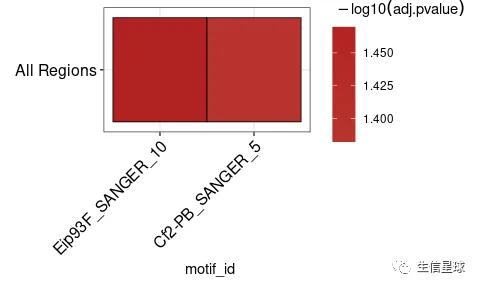
ame_results <- runAme(sequences, control = "shuffle", evalue_report_threshold = 30)
ame_results
#> # A tibble: 2 x 17
#> rank motif_db motif_id motif_alt_id consensus pvalue adj.pvalue evalue tests
#> <int> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl> <int>
#> 1 1 /usr/lo… Eip93F_… Eip93F ACWSCCRA… 5.14e-4 0.0339 18.8 67
#> 2 2 /usr/lo… Cf2-PB_… Cf2 CSSHNKDT… 1.57e-3 0.0415 23.1 27
#> # … with 8 more variables: fasta_max <dbl>, pos <int>, neg <int>,
#> # pwm_min <dbl>, tp <int>, tp_percent <dbl>, fp <int>, fp_percent <dbl>
# 可视化结果
ame_results %>%
plot_ame_heatmap()
FIMO分析motif出现的位置
MotifDb的使用方法: https://bioconductor.org/packages/release/bioc/vignettes/MotifDb/inst/doc/MotifDb.html 以及 https://rdrr.io/bioc/MotifDb/man/MotifDb.html
# MotifDb
# The matrices are obtained from six public sources:
FlyFactorSurvey: 614
hPDI: 437
JASPAR_CORE: 459
jolma2013: 843
ScerTF: 196
stamlab: 683
UniPROBE: 380
cisbp 1.02 874
# Representing primarily five organsisms (and 49 total)
Hsapiens: 2328
Dmelanogaster: 1008
Scerevisiae: 701
Mmusculus: 660
Athaliana: 160
Celegans: 44
other: 177
继续看FIMO的使用:
# Query MotifDb for a motif
e93_motif <- MotifDb::query(MotifDb::MotifDb, "Eip93F") %>%
universalmotif::convert_motifs()
# Scan for the E93 motif within given sequences
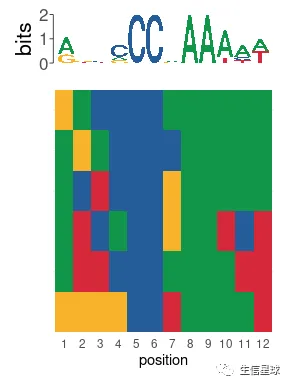
fimo_results <- runFimo(sequences, e93_motif, thresh = 1e-3)
# 可视化
plot_sequence_heatmap(fimo_results$matched_sequence)
还支持导入MEME网页工具的结果

最后,工具绝对不止一种
比如motif分析,http://veda.cs.uiuc.edu/MET/links.html这里列出了一些:
另外,常用的homer也支持,例如之前技能树发布的: Homer软件的介绍-最全面而详细的找motif教程
# 安装方法1:
conda install -c bioconda homer
# 这个路径需要自己修改
perl ~/miniconda3/pkgs/homer-4.10-pl526hc9558a2_0/share/homer-4.10-0/configureHomer.pl -install hg19
# 最后数据下载到
ls ~/miniconda3/pkgs/homer-4.10-pl526hc9558a2_0/share/homer-4.10-0/data/
## 安装方法2:
cd ~/biosoft
mkdir homer && cd homer
wget http://homer.salk.edu/homer/configureHomer.pl
perl configureHomer.pl -install hg19
# 准备数据: 第一列: 染色体; 第二列: 起始位置; 第三列: 终止位置; 第四列: 链的方向
awk '{print $4" "$1" "$2" "$3" +"}' macs_peaks.bed >sample_homer.bed
# 运行(在基因组区域中寻找富集Motifs)
findMotifsGenome.pl sample_homer.bed hg19 sample_homer_motifDir -len 8,10,12
# 参数解释
# -输入文件:awk处理好的Homer Peak/Positions file
# -参考基因组:这里是hg19
# -输出文件:给一个路径和输出文件的名字
# -len:motif大小设置,默认8,10,12;越大需要的计算资源越多
哈佛的课程选择了MEME的DREME工具:https://hbctraining.github.io/Intro-to-ChIPseq/lessons/12_functional_analysis.html
DREME is a motif discovery algorithm designed to find short, core DNA-binding motifs of eukaryotic transcription factors and is optimized to handle large ChIP-seq data sets. DREME is tailored to eukaryotic data by focusing on short motifs (4 to 8 nucleotides) encompassing the DNA-binding region of most eukaryotic monomeric transcription factors.
现在MEME更新了,用STREME 取代了之前的DREME
还有一个MEME-ChIP :
MEME-ChIP performs DREME and Tomtom analysis in addition to using tools to assess which motifs are most centrally enriched (motifs should be centered in the peaks) and to combine related motifs into similarity clusters. It is able to identify longer motifs < 30bp, but takes much longer to run.
最近还发布了一个不基于peaks的工具:NoPeak: k-mer-based motif discovery in ChIP-Seq data without peak calling (Bioinformatics, btaa845, https://doi.org/10.1093/bioinformatics/btaa845, 29 September 2020)
Here, we present a novel approach to reliably identify transcription factor-binding motifs from ChIP-Seq data without peak detection. By evaluating the distributions of sequencing reads around the different k-mers in the genome, we are able to identify binding motifs in ChIP-Seq data that yield no results in traditional pipelines.
还有其他基于R的工具:
rGADEM (motif discovery): http://bioconductor.org/packages/devel/bioc/html/rGADEM.html
MotIV (motif validation): http://bioconductor.org/packages/devel/bioc/html/MotIV.html