217-重温ChIP-Seq相关的数据格式
刘小泽写于2020.12.01 重新看了下哈佛之前的ChIP-Seq课程,感觉其中介绍的比较系统,数据格式部分值得拿出来重新看一下 官方资料在:https://hbctraining.github.io/Intro-to-ChIPseq/lectures/Fileformats.pdf
首先是NGS分析的大体流程
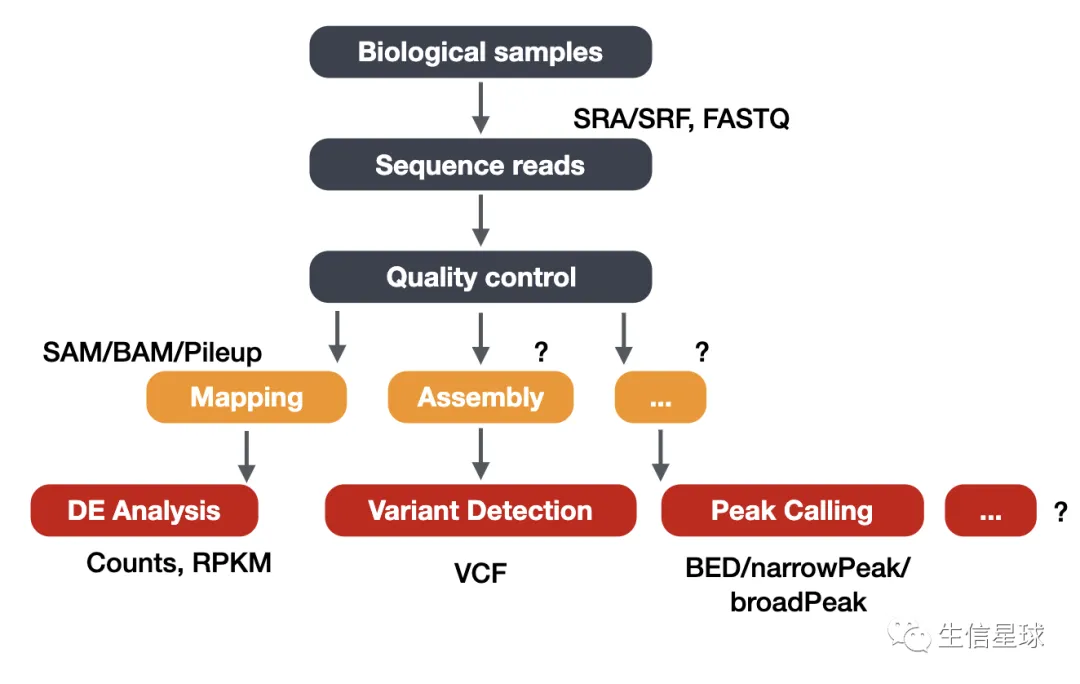
其中ChIP-Seq主要会与peaks打交道,它的本质是一个BED文件;另外还有覆盖度相关的bw文件
常用的文件
主要有三大类:
- 测序数据:fasta、fastq
- 比对数据:sam、bam
- 记录基因组特征数据:bed、wiggle、gtf、gff
其中有一些是二进制类型(如bam、bigwig等),是无法直接查看的
前两类比较好理解,第三种类型中比较特殊一点
基因组特征数据的几个特点
一般是tab分割
包含的信息都是基于基因组坐标的(都记录了染色体、起始终止)
有的文件是1-based,有的是0-based
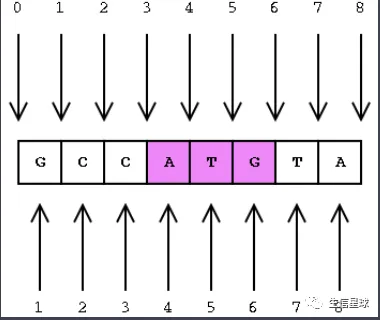
关于1-based和0-based需要提一下
正如下图,上面一行数字是0-based,表示从0开始计数,是程序员的最爱;而下面的是1-based,是生物学家的最爱。关于紫色的ATG,双方有不同的表示方法
- 0-based:ATG位置是(3,6] ,它的长度就是6-3=3
- 1-based:ATG位置是[4,6],它的长度是6-4+1=3
- 因此可以看到,1-based需要用end-start后再加上1才是真实的长度
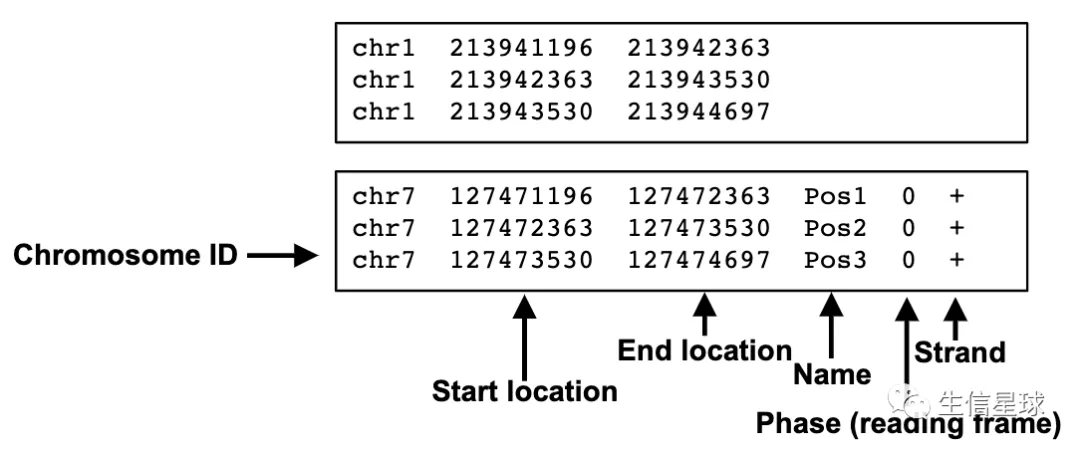
基因组特征数据中的BED文件
可以是tab或者空格分隔
它是0-based
前三列数据必须是:染色体、起始坐标、终止坐标
其余还有9列是可选的,除了3列的还有常见的6列BED
基因组特征数据中的BedGraph文件
主要是用于基因组浏览器显示连续的数值变化(比如看测序数据的基因组覆盖度)
它也是基于BED格式,因此也是0-based。但特殊之处在于它的score列是第4列,而非第5列;另外它还必须包含track line(这个在平常的BED中不是必须的)
从图中也可以看到,它记录了全部的详细数据,并没有对数据进行“压缩”。其中的第4列data value可以是整数,可以是正或负值

如何生成bedGraph呢?
# -d参数就是产生depth文件,-bg产生bedgraph文件 bedtools genomecov -ibam input.bam -bg > depth.bedgraph
基因组特征数据中的Wiggle文件
和bedgraph类似,但这个数据是经过“压缩”的。意思就是它感觉bedGraph数据信息虽然全,但也导致了文件比较大。于是它想了个办法,在保留原有信息同时,对数据进行改造,比如下面这两种都是常见的方法
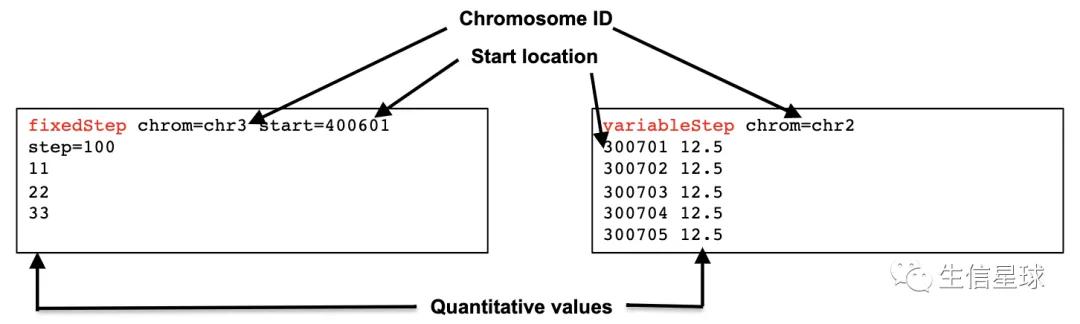
一种是规定步长,比如step=100,就是每隔100个bp统计一下,中间有多少reads
另一种是直接记录,1-100直接有多少reads,101-200直接又有多少reads
特殊之处在于:它是1-based
基因组特征数据中的bigwig文件
是传承于wiggle二进制文件格式,特点是读取很快,并且文件比较小
1-based格式
之前bw都是需要通过wiggle生成的,不过现在可以通过bedGraph直接得到。比如
# install conda install -c bioconda ucsc-bedgraphtobigwig # bedgraph to bigwig for i in `ls *bedgraph` do (head -n 1 $i && tail -n +2 $i | sort -k1,1 -k2,2n | awk '{print $1,$2,$3,$4}' OFS="\t" ) > $i.sort && bedGraphToBigWig $i.sort hg19.chrom.sizes $i.bw done
直观展示文件大小
#如果一个 bam是 2.7G
# 那么
# depth 55G
# begraph 550M
# bigwig 15M
# bigwig最小,通常会用取平均值等方法来表示一个窗口内所有碱基的测序深度,信息是稍微有点失真的,但是窗口相比染色体而言非常的小,但是直观判断是没问题的